기사본문
[BioS 레터]암세포서 PD-L1 높은 이유는 뭘까?
입력 2021-08-19 09:46 수정 2024-08-19 10:50
정다혜
암을 정복하기 위해 수많은 기술과 연구가 시행되고 있지만 우리는 여전히 암과의 사투를 벌이고 있다. 암이란 무엇일까? 초창기에는 단순하게 죽어야 할 세포가 죽지않고 계속 증식되어서 생긴 덩어리라고 여겨졌다. 그래서 1세대의 암 치료요법으로 항암제와 절제 수술법을 통해 암을 제거했다. 하지만 대다수의 암 환자들이 치료되지 못했다. 추후의 연구를 통해서 암세포는 유전자 돌연변이를 통해서 자신이 살아남기에 유리한 조건들을 만들고 있는 것이 밝혀졌다.
우리 몸은 항상성을 위해 새로운 세포가 만들어지면 그만큼 제거가 되어야 하는데 암을 유발하는 유전자가 발현되면서 죽지않고 계속 증식이 되는 것이다. 이렇게 암을 유발할 가능성을 가진 유전자를 ‘Oncogene’이라고 했다. 대표적으로 ‘c-Myc’이라는 Oncogene이 높게 발현하면 ‘세포 증식(Cell proliferation)’이 유도되어 빠르게 암세포로 증식될 수 있다[1]. 그래서 2세대의 암 치료 요법으로 이러한 Oncogene을 타깃하는 유전자 치료법들이 등장했다. 하지만 유전자를 타깃하기 위해서는 바이러스 감염을 통한 방법이 주를 이루는데 이는 외부의 물질을 주입하다 보니 여러 문제점이 생겼다.
암 연구가 계속 이루어지면서 2013년 ‘Immunity’에 ‘Oncology Meets Immunology: The Cancer-Immunity Cycle’이라는 논문이 발표되었고, 암세포가 ‘면역세포’를 통해 제거될 수 있는 것에 주목하였다. 이 논문에서는 암과 면역세포 사이의 사이클을 7개의 순서로 설명한다. 먼저 ①암세포가 사멸하면서 ‘항원(Antigen)’을 방출하고, ②‘수지상 세포(Dendritic cells(DCs)’가 항원을 잡아먹고 항원 일부를 자신의 표면에 제시한 상태로 림프절로 이동한다. 여기서 ③DCs의 표면에 있는 항원을 T세포가 인지하고 기억한 상태로 활성화가 되어 ④항원이 방출된 암이 있는 곳으로 간다. ⑤그리고 암 조직 사이로 침투하여, ⑥자신이 기억하고 있는 항원을 제시하고 있는 암을 T세포가 인지하게 된다. 그리고 ⑦T세포가 암을 제거한다[2][그림 1]. 이 사이클에서 가장 핵심적인 것은 바로 T세포이기 때문에 분자수준에서 면역 메커니즘을 이해하여 T세포의 기능을 강화하기 위한 3세대 항암요법인 ‘면역항암요법’이 등장했다(T세포 외에도 NK 세포 등 다양한 면역세포들을 이용한다).
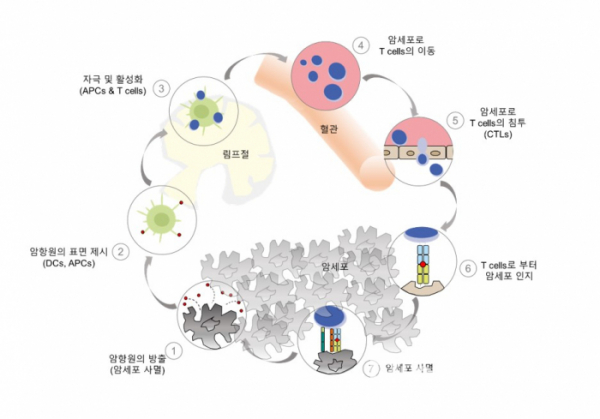
▲그림 1. The Cancer-Immunity Cycle
면역세포의 기능 강화 – 면역관문억제제
사이클의 3번째 단계에서 T세포가 활성화되면서 항원을 인지할 수 있는 T세포 수용체(T cell receptor, TCR)가 만들어진다. 그래서 암 근처로 이동하였을 때 암에 나타난 항원을 이 수용체를 통해 먼저 결합할 수 있다. 그리고서 ‘공동자극 분자(Co-stimulator molecule)’가 추가로 발현하여 암세포에 결합하고 암세포를 제거하는 ‘Granzyme B’와 같은 물질들을 방출하여 ‘세포자살(Apoposis)’을 일으켜 제거한다[3]. 하지만 이러한 물질이 계속 방출되면 정상적인 세포도 공격하여 ‘자가면역질환’이 생길 수 있으므로 T세포에 ‘공동억제 분자(Co-inhibitory molecule)’가 발현해서 활성을 멈춘다[4]. 이때 특정 암세포는 이러한 작용을 교묘히 이용해서 공동억제 분자와 결합할 수 있는 분자를 많이 발현하는 특징을 갖는다. 그렇게 되면 T세포가 암세포를 인지해도 암세포를 제거하는 물질을 방출할 수가 없게 된다[5].
2018년 공동억제 분자와 T세포의 결합을 억제함으로써 T세포의 활성화를 끌어내는 일명 ‘면역관문억제제 (Immune checkpoint Blockade, ICB)’라는 개념을 규명한 미국의 제임스 P. 앨리슨과 일본의 혼조 타스쿠가 노벨생리의학상을 받았다. 이러한 면역체크포인트 단백질로는 대표적으로 ‘Programmed death ligand 1(PD-L1)’이 있는데 T세포에 발현된 ‘Programmed death 1(PD-1)’과 결합한다. 특히 혼조 타스쿠는 이 PD-1의 억제를 통해 기존의 표적항암제와 달리 전이암 환자들에서 극적인 완화 및 치료를 확인하였다.
PD-L1의 발현 메카니즘과 적용
PD-1 억제라는 뛰어난 발견과 효능에도 일부 암종(흑색종, 비소세포 폐암 등)에서만 효과를 나타내는 한계를 가졌다. 이러한 한계를 극복하기 위해 대만의 미엔치엔 헝 팀은 암세포에서 PD-L1의 발현이 높은 이유를 먼저 확인하고자 하였다. 특히 유방암의 서브 타입 중 하나인 ‘삼중음성유방암(Triple-negative breast cancer, TNBC)’은 다른 서브 타입에 비해 성장 속도가 빨라 상당히 공격적인 암종으로 알려졌다. 또한 PD-L1이 과발현되어 있다고 알려졌지만 면역관문억제제를 통한 반응률은 15~20%에 그친다. 이러한 삼중 음성 유방암의 치료를 목적으로 헝 팀은 면역관문억제제 발현 기작 연구를 통해 효과적인 치료 방법을 모색하고자 하였다.
번역 후 변형(Post-translational modification, PTM)
먼저 이들은 PD-L1의 성질을 파악하여 2016년, ‘Nature communication(IF 12.1)에 ‘Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity’라는제목의 논문을 게재했다. PD-L1은 PD-1과 결합하기 위해 세포막에 위치하는 막단백질이다. 세포막에 자리를 잡기 위해 대부분 막단백질은 ‘번역 후 변형(Post-translational modification, PTM)’ 과정을 겪고 이때 단백질들은 역동적인 성질을 갖게 된다. 번역 후 변형과정은 중심원리(Central dogma) 이후의 단백질에 추가로 일어나는 과정이다.
번역 후 변형과정 중 하나로 ‘당화(Glycosylation)’가 있는데 이것은 말 그대로 단백질에 ‘당(Sugar)’이 붙는 것이고, 우리는 이것을 ‘당단백질 (Glycoprotein)’이라 한다[6][7]. 당이 단백질에 합성되는 것이 왜 중요할까? 일반적으로 단백질에 당이 붙는 것을 통해 단백질이 ‘안정화’된다고 알려져 있다. 단백질이 ‘안정화’가 되는 것은 어떤 것을 의미할까? 단순하게 생각하면 단백질이 사라지지 않고 계속 남아있을 수 있다는 뜻이다. 우리 몸은 항상성을 유지하기 위해 단백질이 생성되고 사라지고, 다시 생성되고 사라지는 것을 반복한다. 각각의 단백질들은 자신의 ‘라이프 타임(Life time)’을 가지고 있다. 만약 암세포와 같은 환경에서 특정 단백질이 자신의 라이프 타임 이상으로 머물러 있게 되면 이것은 특정 단백질이 안정화되었다고 말할 수 있다. 논문에서는 암세포에서 PD-L1이 높게 발현하는 이유가 바로 당화를 통한 안정화때문이라는 것을 밝혀냈다.
유비퀴틴-프로테아좀 시스템(Ubiquitin-Proteasome System, UPS)
그렇다면 안정화를 갖기 위해 PD-L1은 어떻게 조절되었을까? 일반적으로 단백질이 사라지기 위해서는 일련의 과정을 거치게 된다. 여러 방식이 있겠지만 대표적으로 ‘유비퀴틴-프로테아좀 시스템(Ubiquitin-Proteasome System, UPS)’이 있다. 간단하게 ‘유비퀴틴(Ubiquitin, Ub)’이라는 물질이 제거하고 싶은 단백질에 붙으면, 이것을 믹서기와 같은 ‘프로테아좀(Proteasome)’이 인지해서 제거하는 것이다.
유비퀴틴은 76개 정도의 아미노산으로 이루어진 단백질이고, 타깃 단백질에 추가되는 과정을 ‘유비퀴틸화(Ubiquitylation)’라 한다. 이 과정은 세 종류의 E1(Ubiquitin-activating enzyme), E2(ubiquitin-conjugating enzyme) 그리고 E3(ubiquitinligase)라고 하는 단백질의 도움으로 차례대로 수행된다. 유비퀴틴의 C-말단에 ‘글라이신(Glycine)’ 잔기와 타깃 단백질의 ‘라이신(Lysine)’ 잔기가 결합할 수가 있는데 이 과정에서 삼총사 단백질이 사용된다. E1을 통해 유비퀴틴이 활성화가 되고, E2가 활성화된 유비퀴틴을 받아 E3를 통해 글라이신과 라이신 잔기가 결합하게 한다. 유비퀴틴은 6개의 라이신 잔기를 가지고 있으므로 E1에 의해 새롭게 활성화된 유비퀴틴의 글라이신과 타깃 단백질에 붙어있는 유비퀴틴의 라이신과 결합할 수 있다. 이런 과정의 반복으로 유비퀴틴이 여러 개가 연결될 수 있고, 우리는 이를 ‘폴리 유비퀴틴 체인(Poly ubiquitin chain)’이라고 한다. 이것이 형성되면 프로테아좀에 의해 인지되고, 단백질은 제거된다[8].
이제 다시 PD-L1으로 돌아가보자. PD-L1은 당화를 통해 안정화를 얻게된다고 하였다. 간단히 말하면 당화로 인해서 E3가 구조적으로 타깃 단백질에 결합할 수 없게 된다. 그러면 유비퀴틸화가 일어날 수 없게 되고 프로테아좀으로 제거가 되지않게 되기 때문에 안정성을 얻게 된다.
N-glycosylation
그렇다면 이러한 당화는 어떻게 일어날까? 당화는 ‘소포체(Endoplasmic reticulum, ER)’에서 시작된다. `핵(Nuclear)’에서 `번역(Translation)’되어 나온 초기의 단백질 형태인 ‘폴리펩타이드(Polypeptide)’가 소포체로 들어갈 때, `막 관련 올리고당 전이효소 복합체(Membrane-associated oligosaccharyl transferase complex, OST)’에 의해 14개의 당이 결합한다[9]. 이때 폴리펩타이드의 ‘아스파라진(Asparagine, Asn, N)’에 결합하기 때문에 ‘N-당화(N-glycosylation)’ 과정이라 한다. 아스파라진에 당이 붙으면서 단백질 ‘접힘(Folding)’이 일어나고, 이때 접힘이나 당화가 제대로 이루어지지 못한 단백질은 소포체 내에서 이것을 인지한다. 그리고 위에 우리가 언급한 유비퀴틴-프로테아좀 시스템을 이용하여 제거한다. 이 과정은 소포체에서 인지해서 소포체 밖으로 내보내서 ‘세포질(Cytosol)’에서 일어난다. 이 과정을 ‘소포체 관련 단백질 분해경로(Endoplasmic-reticulum-associated protein degradation, ERAD pathway)’라 한다[10]. 접힘과 당화가 잘 형성된 단백질은 ‘골지체(Golgi apparatus)’로 이동해서 당의 개수들이 조절되면서 우리가 알고있는 기능들을 할 수 있는 단백질의 모습을 갖춘다.
헝 팀은 당화가 일어나는 아스파라진 위치를 잘라낼 수 있는 효소인 ‘PNGaseF’와 당화가 처음 일어나는 과정을 막을 수 있는 ‘Tunicamycin(TM)’을 통해 PD-L1이 당화가 되어있다는 것과 이러한 효소와 약물이 존재할 때 유비퀴틴의 양이 증가하는 것을 통해 당화가 PD-L1의 안정화를 유도하는 것을 확인하였다. 추가로 LC-MS/MS 분석을 통해 N192, N200 그리고 N219에서 당화가 일어나는 것을 확인하였다.
GSK3β의 활성화를 통한 유비퀴틴-프로테아좀 시스템의 유도
다음으로 그렇다면 이런 과정이 어떻게 일어나는지 알기 위해 조사하는 과정에서 이들은 ‘GSK3β’라는 ‘카이네이즈(Kinase)’를 발견하였다. 카이네이즈는 단백질에 ‘인산화(Phosphorylation)’를 시키는 단백질이다[11]. GSK3β로 인해 인산화된 단백질은 E3 ligase 중 하나인 ‘β-TrCP’에 의해 인지되어 폴리 유비퀴틴이 붙어 프로테아좀으로 제거된다. 헝 팀은 따라서 GSK3β를 활성화할 수 있는 메커니즘을 확인하고자 하였다. 일반적으로 GSK3β는 ‘표피성장인자(Epidermal Growth Factor, EGF)’, ‘인슐린 성 성장인자(Insulin-like growth factor-1)’ 그리고 ‘전환성장인자(Transforming growth factor(TGF-β)’와 같은 ‘성장인자(Growth factor)’들에 의해 비활성화된다고 알려져 있다. 따라서 이들은 다양한 암세포 주에 성장인자들을 처리하였고, EGF에서 PD-L1이 높게 발현되는 것을 확인하였다.
또한 EGF는 ‘표피성장인자수용체(Epidermal Growth Factor Receptor, EGFR)’와 결합하여 신호전달을 할 수 있다. 따라서 헝 팀은 EGFR에 결합할 수 있는 ‘Epiregulin’, ‘TGF α’와 같은 인자들을 처리했고, 이때 역시 PD-L1의 증가를 확인할 수 있었다. GSK3β는 ‘세린(Serine)’ 9번째 자리가 인산화가 되면 비활성화되는 것으로 알려져 있는데 EGFR이 활성화되었을 때 GSK3β의 세린 9번의 인산화를 Western blot실험을 통해 확인하였다. 이러한 결과들을 통해 EGFR이 활성화가 될 때 GSK3β가 비활성화되어 PD-L1의 안정화가 증가하는 것을 밝혔다[그림 2]. 추가적으로 EGFR을 억제할 수 있는 ‘Gefitinib’ 항체를 사용하였을 때 GSK3β가 활성화를 통한 PD-L1의 발현이 낮아져 BALB/c 마우스 모델에서도 암의 크기가 감소하는 것을 확인하였다.
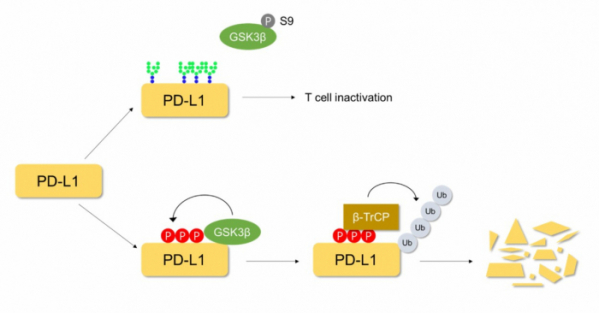
▲그림 2. EGF mediated PD-L1 stabilization contributing to T cell immune escape
PD-L1의 당화 성질을 이용한 암 치료요법 적용
PD-L1의 당화를 통한 안정화와 이것을 억제하였을 때 암 치료에 효과적인 전략이 될 수 있는 것을 말해주는 것을 시작으로 2018년 ‘Molecular Cell (IF 14.5)’에 ‘Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1’이라는 논문을 발표됐다. 이 논문에서는 ‘Metformin’이라는 당뇨병 환자 치료에 쓰이는 약물을 통해 PD-L1의 인산화가 유도되어 소포체 내에서 당화가 제대로 이루어지지 못해 ERAD를 통해 제거되는 암 치료요법 전략을 설명하고 있다.
그리고 같은해 ‘Cancer cell (IF 23.9)’에 ‘Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1’이라는 논문이 게재되었다. PD-L1에 생기는 당화가 PD-L1 자체의 안정성에 이바지하는 것 이상으로 T세포의 PD-1과 결합하기 위한 구조적인 영향을 준다는 것을 밝혔다. 당화가 일어나지 않은 PD-L1이 높게 발현하는 세포에서는 실제로 PD-1과의 결합이 낮은 것을 확인할 수 있었다. 또한 T세포의 반응이 증가하고, BALB/c 마우스 모델에서 역시 암의 크기가 감소하는 것을 보았다. 그들은 이러한 현상이 나타나는 것을 골지체에 있는 ‘β-1, 3-N-acetylglucosaminyltransfera 3(B3GNT3)’이라는 단백질이 영향을 준다고 생각했다. 소포체에서 14개의 당이 단백질에 붙는 것을 시작으로 이 과정이 잘 이루어지면 골지체로 넘어가게 되고 골지체에서 당의 개수가 조절되면서 정상적인 단백질의 모습을 갖춘다. B3GNT3는 당을 붙이는 역할을 하는 단백질이다.
헝 팀은 EGF를 통해 B3GNT3의 양이 증가하는 것을 확인했을 뿐만 아니라 PD-L1의 N192와 N200에 영향을 미치는 것을 확인하였다. PD-L1과 PD-1의 결합에 당화의 중요성을 확인한 결과를 토대로 그들은 PD-L1의 당화(N192, N200)와 결합할 수 있는 항체를 만들었다. 기존의 PD-L1 자체를 잡는 항체와는 차별적인 전략이었다. 항체의 이름은 ‘STM 108’로 하였고, 일반적으로 ‘Antibody-Drug Conjugate(ADC)’에 사용되는 약물인 ‘Momomethyl auristatin E(MMAE)’를 달았다. ADC의 기술의 관건은 ADC가 타깃하는 단백질과 함께 ‘내재화(Internalization)’되어 ‘리소좀(Lysosome)’에 의해 분해되어 약물이 암세포에 잘 전달되어야 한다. 내재화 과정에는 크게 ‘Clathrin-mediated endocytosis(CME)’과 ‘Caveolae-dependent endocysotis(CDE)’ 두 과정이 잘 알려져 있다. 헝 팀은 CME와 CDE 억제 약물을 통해 STM 108이 CDE 과정을 통해 내재화되는 것을 확인하였고, 마우스 모델을 통해서도 40일에 죽던 마우스가 80일 이상 생존함을 통해 항체에 대한 높은 효과를 보여주었다.
헝 팀의 논문들을 통해 단순한 바이오 마커의 타깃이 아닌 그 바이오 마커의 메커니즘 이해를 통한 치료방법은 흥미로울 뿐 아니라 앞으로 계속 시행해나가야 할 연구과정이라고 생각한다. STM 108의 마우스 이상의 효능은 아직 확인하지 못하였지만 이러한 깊은 연구들이 지속된다면 좋은 결과가 나오리라고 기대해본다.
Reference
[1] Donald M. Miller, c-Myc and Cancer Metabolism, Clin Cancer Res, 2012; 10.1158/1078-0432.CCR-12-0977
[2] Daniel S. Chen Oncology Meets Immunology: The Cancer-Immunity Cycle, Immunity, 2013; https://doi.org/10.1016/j.immuni.2013.07.012
[3] Ilona rousalova, Granzyme B-induced apoptosis in cancer cells and its regulation, International journal of oncology, 2010; 10.3892/ijo_00000788
[4] Norio Chihara, Induction and transcriptional regulation of the co-inhibitory gene module in T cells, Nature, 2018;
[5] Ostrand-Rosenberg S, Horn LA, Haile ST. The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity. J Immunol. 2014;193(8):3835-3841. doi:10.4049/jimmunol.1401572
[6] Li, C.-W. et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun, 2016; 7:12632 doi: 10.1038/ncomms12632
[7] Jung-Mao Hsu, Posttran Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy, Cancer research, 2018; 10.1158/0008-5472.CAN-18-1892
[8] Nico P. Dantuma, The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution, 2014; https://doi.org/10.3389/fnmol.2014.00070
[9] Xu C., Glycosylation-directed quality control of protein folding, Nat. Rev. Mol. Cell Biol. 2015; 16: 742-752
[10] Ferris S.P, Glycoprotein folding and quality-control mechanisms in protein-folding diseases, Dis. Model. Mech. 2014; 7: 331-341
[11] Doble, B. W. & Woodgett, J. R. GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 (2003).







