기사본문
[남궁석의 신약연구史]'알파폴드2'와 단백질디자인
입력 2022-02-17 14:29 수정 2022-02-17 14:29
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
알파폴드2의 놀라운 구조예측 능력은 과학자뿐만 아니라 대중들의 많은 관심을 모았다. 구조생물학자들은 아미노산 서열의 진화 정보를 이용하여 단백질의 3차구조를 매우 정확히 규명하는 방법이 나왔다는 사실과, 이를 이용하여 현재 구조생물학의 화두인 세포 내의 거대 고분자 복합체의 구조와 작동원리를 좀 더 효율적으로 푸는 것에 관심을 가졌다면, 산업계의 연구자들이나 대중들은 정확한 단백질 구조예측이 우리에게 어떤 실용적인 가치를 창출할지에 대해서 더 관심을 가졌다. 구조예측이 신약개발에 어떤 영향을 줄지에 대한 관심이 높아졌다. 일부 매체에서는 알파폴드2의 단백질 구조예측이 바로 수많은 신약을 탄생시킬 수 있는 시금석처럼 묘사되기도 하였다.
그러나 현대의 신약개발에서 단백질의 구조정보, 특히 소분자 물질과 표적 단백질과의 결합구조가 신약개발에서 필수불가결한 정보가 되긴 했지만, 단백질 구조규명(혹은 정확한 예측)은 신약개발에 있어서 필요한 한가지 정보일 뿐이다. 기존에 구조를 모르던 단백질의 구조가 실험적인 방법, 혹은 예측에 의해서 밝혀져도 이것이 바로 신약개발과 직결되지는 않는다.
오히려 아직까지 타깃 단백질의 구조가 규명되지 않았던 경우에도(구조가 있는 경우에 비해서 어려움을 겪긴 하겠지만) 많은 신약들이 개발되어 왔다는 것을 생각하면 단백질의 구조를 정확히 예측하는 것과 신약개발과의 간극은 상당하다. 단백질 구조예측을 다루는 이번 연재의 마지막 회에서는 현재의 알파폴드2 등을 위시한 여러가지 예측 기술들이 어떤식으로 신약개발을 도울 수 있는지, 그리고 앞으로 보다 효율적인 신약개발을 위해서 발전의 여지가 있는 분야는 어떤 부분일지에 대해서 알아보도록 한다. 일단 현대 신약의 두가지 부류라고 할 수 있는 소분자 기반의 신약과 단백질 기반 신약으로 나누어 설명하고자 한다.
단백질 구조예측과 소분자 기반 신약개발
알파폴드2 등의 단백질 구조예측이 직접적으로 가장 큰 영향을 줄 분야는 아무래도 단백질에 결합하는 소분자 물질기반의 신약개발일 것이다.[1] 단백질 구조규명이 일반화된 현재에 와서 어떤 단백질에 결합하는 약물을 개발할 때 흔히 채용되는 방법은 구조기반 신약개발(Structure-based drug development, SBDD)이다. 약물의 타깃이 되는 단백질 구조가 존재하면 약물이 어떻게 단백질과 결합하는지의 결합모드를 파악하기 위하여 두가지 방법론이 존재하는데, 한가지는 약물과 단백질과의 결합구조를 실험적으로 규명하는 것이고, 다른 방법은 아직 결합구조가 규명되지 않은 약물과 단백질의 결합구조를 예측하는 것이다. 후자의 방법론은 통칭 '도킹(Docking)'이라고 불리며, 약물과 단백질 구조의 물리적인 성질에 근거하여 단백질에 어떻게 약물이 결합할 수 있는지를 예측하는 것이다. 이 두가지 방법은 독립적이기 보다는 같이 사용된다. 일단 어떤 단백질에 결합하는 선도 화합물(Lead compounds)과 단백질의 결합구조가 규명되면, 좀 더 좋은 활성을 가지도록 선도 화합물과 단백질간의 구조에 기반하여 화합물을 디자인하여 다시 복합체 모델링 및 구조규명을 하고, 이를 통하여 더 좋은 결합활성을 가지는 화합물을 디자인하는 것이 일반적인 일이다.
그렇다면 단백질 구조예측이 이러한 과정을 어떻게 도와줄 수 있을까? 만약 실험적인 방법으로 구조를 규명하기 힘든 단백질에 대한 약물을 개발하는 경우라면 단백질의 구조예측이 큰 도움이 된다. 구조예측의 정확도가 낮았던 과거에는 대략적인 구조를 예측하더라도 세부적인 아미노산 잔기의 방향까지는 정확한 예측이 불가능했고, 실제 단백질 구조와 예측된 구조와의 차이는 도킹 등의 가상 스크리닝의 정확도를 현저히 떨어뜨렸다. 그러나 알파폴드2의 예측모델은 거의 실험적인 구조에 필적하는 정확도를 보여주고 아미노산의 잔기의 방향까지 거의 정확히 예측하고 있으므로 알파폴드2에서 예측된 단백질 구조모델은 실험적으로 규명된 구조와 큰 차이 없이 가상 스크리닝에 이용될 수 있을 것으로 보인다.
물론 구조예측으로 얻어진 단백질 구조를 이용하여 소분자 기반 신약을 개발하는데는 분명히 한계가 존재한다. 이 중 하나는 많은 약물 타깃 단백질은 여러가지 구조적인 상태를 가지는 경우가 있고, 약물은 어떤 특정한 상태에만 결합하는 경우가 있다. 가령 만성 골수성백혈병의 원인 단백질인 BCR-ABL 인산화효소는 활성화된 상태와 비활성화된 상태가 서로 다른 구조를 가지고 있고, 여기에 특이적인 치료제인 '글리벡(Gleevec, imatinib)'은 BCR-ABL 인산화효소의 비활성화된 형태에만 결합하게 된다.[1] 그러나 알파폴드2 등에서 예측된 구조는 어떤 단백질이 가질 수 있는 여러가지 상태 중에서 구조 데이터베이스 등에 가장 많이 분포하는 형태와 유사한 구조일 가능성이 높다. 즉, 예측된 단백질의 구조가 실제로 어떤 약물이 결합하는 구조와는 다른 형태라서, 화합물과 단백질간의 도킹을 제대로 수행할 수 없는 경우도 존재한다. 그리고 단백질의 구조 자체가 이에 결합하는 화합물과의 결합에 의해서 미세하게 변화하는 경우도 생기는데, 이러한 것을 현재의 구조예측으로는 쉽게 해결할 수 없다는 것이 문제이다.
따라서 소분자 물질을 이용한 신약개발에서는 단순히 구조를 모르는 단백질의 구조예측만으로는 충분하지 않으며, 단백질과 약물간의 결합 구조를 정확하게 예측하고, 나아가서 단백질과 약물과의 결합력을 정확히 예측하는 것이 필요하다. 이러한 예측은 단백질 구조를 정확하게 예측하기 이전부터 단백질과 약물 간의 물리적인 성질을 감안한 여러가지 예측 방법들이 개발되어 왔고, 실제로 이를 이용하여 많은 약물들이 개발되어 왔다. 그러나 이러한 예측은 아직도 실험적인 방법에 비해서 상당한 오차를 가지고 있으며, 주로 실험적인 검증을 거칠 후보 화합물의 수를 줄이는 1차 스크리닝 과정에서 많이 사용되어 왔다. 즉, 모든 과정을 예측과 계산만으로 신약을 개발하기에는 아직까지는 많은 한계를 가지고 있는 셈이다. 그러나 딥마인드가 인공지능 기법을 이용하여 단백질 구조예측을 혁신시킨 것과 비슷한 수준의 진보를 단백질과 약물의 결합 방식의 예측 그리고 이의 물리적 특성의 예측에서 이룩할 수 있다면 신약개발 과정에서의 진정한 혁신이 일어날 것으로 생각된다.
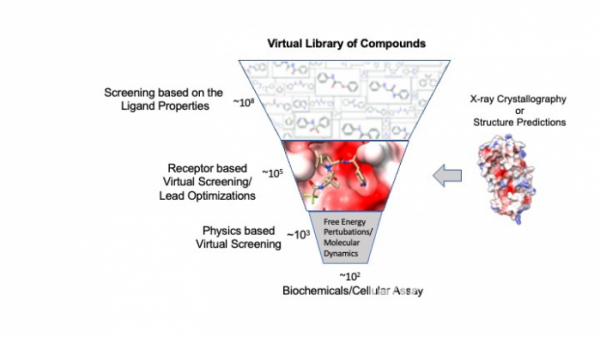
▲소분자 신약후보물질의 구조기반 탐색. 현재 소분자 신약후보물질의 개발에 있어 주도적인 패러다임인 구조기반 신약탐색(Structure-Based Drug Discovery)은 신약의 타깃이 되는 단백질의 구조를 기반으로 여기에 결합하는 화합물을 방대한 크기의 가상 화합물 라이브러리에서 실험으로 활성을 검정할 수 있는 수준으로 줄여주는 역할을 주로 수행한다. 구조 예측은 만약 타깃이 되는 단백질의 구조가 알려지지 않았을 경우, 여기에 사용될 타깃 구조를 제공해 주는 역할로 유용하게 사용될 것이다. 단백질의 구조에 대한 정보는 신약개발의 극히 일부 과정인 신약 후보물질의 도출에 꼭 필요한 정보 중의 하나이긴 하지만, 신약개발에 필요한 여러가지 난관 중에서 극히 일부에 불과하다.
단백질 기반의 신약개발과 구조예측
현대의 신약개발에서 소분자 물질과 함께 중요한 한 축을 차지하고 있는 것이 단백질 기반의 신약개발이다. 그렇다면 단백질 기반의 신약개발에서 구조예측은 어떤 역할을 할까.
일단 소분자 물질의 경우 표적 단백질과의 결합 활성을 약물로 사용할 정도로 올리기 위해서는 소분자 물질과 단백질과의 결합 방식을 구조규명(혹은 구조예측)을 통하여 수행하는 것이 필수적이지만, 단백질 기반의 신약의 대표적인 형태인 항체 등의 경우 단일항원 항체 선별, 파지 디스플레이 등의 방법으로 높은 결합력을 가진 항체를 선별하는 데 구조정보가 꼭 필수적인 것은 아니다. 물론 표적 단백질과 항체와의 복합체 구조 정보가 존재한다면 어느 정도 결합력이 있는 항체를 좀 더 최적화하는데 보탬이 될 것이다. 그러나 단백질 간의 복합체의 구조를 결정하는 것은 소분자 물질과 단백질의 구조를 결정하는 것보다 훨씬 고난이도의 일이라는 것이 걸림돌이다.
결국 구조예측이 단백질 기반의 신약을 개발하는데 도움이 되려면 표적 단백질과 항체 등의 단백질 신약 간의 단백질 상호작용을 정확하게 예측하는 것이 필요하다. 그러나 현존하는 예측 방법론은 알파폴드2에서 단백질의 구조를 예측한 수준으로 높은 정밀도로 단백질간 상호작용을 예측하는데 한계가 있다.
기존의 단백질 상호작용 예측방법은 주로 물리적인 성질에 기반한 것으로써, 서로 상호작용하는 단백질에 대해서 수많은 상태의 결합방식을 탐색한 후 이들의 단백질 복합체의 에너지 상태를 추정한 후, 이를 통하여 에너지적으로 가장 안정된 단백질 결합상태를 추정하게 된다. 이 과정에서 단백질 간의 경계에 존재하고 있는 아미노산들의 방향을 재정렬하여 가장 최적의 에너지 상태를 만드는 과정을 거치게 된다. 결합하는 두 개의 단백질이 구조를 크게 변화하지 않은 상태에서 최적의 상호작용을 찾는(Rigid-body docking) 것이므로 만약 결합하는 한 쪽의 단백질의 구조가 다른 쪽에 의해서 변하는 경우라면 정확한 결합을 찾기가 어렵다는 문제점이 있다. 단백질의 결합면이 변하는 구조를 예측하기 위해서 여러가지 방법이 강구되어 왔지만, 아직도 단백질 간의 상호작용을 예측하는 것은(알파폴드 등장 이전의 단백질 구조예측과 마찬가지로) 쉽지않은 문제이다. 알파폴드2가 구조예측의 능력을 과시한 CASP14와 함께 진행된 단백질 간의 상호작용을 예측하는 대회인 CASP14-CAPRI(Critical Assessment of Prediction of Interactions)의 결과를 살펴보면 아직 단백질 간의 상호작용의 예측은 발전이 필요하다는 것을 알 수 있다.[3] 문제로 주어진 12개의 단백질 복합체 중에서 기존에 유사한 구조가 풀려 있는 두 개의 단백질의 결합구조를 예측하는 것과 같이 비교적 난이도가 낮은 예측은 대부분의 참가자들은 어렵지 않게 상당히 실제 구조와 흡사한 구조를 예측했지만, 기존에 알려진 구조와 상동성이 별로 없는 구조간의 결합, 혹은 여러 개의 단백질로 이루어진 거대 복합체의 예측은 실제 복합체와 그다지 근접한 예측을 하지 못한 편이었다.
알파폴드2의 등장 이후 자연계 내에서 상호작용을 이루는 단백질들의 상호작용 예측이 알파폴드2에 의해서 어느정도 정확히 이루어진다는 보고가 있었다.[4] 알파폴드2의 단백질 구조예측은 단백질 구조를 이루는 상호작용이 진화적으로 보존되어 있다는 원리에 의해서 이루어지며, 따라서 진화적으로 상호작용이 보존되어 있는 단백질 간의 상호작용도 어느 정도 예측할 수 있는 능력이 있다는 것은 크게 놀랄 만한 일은 아니었다. 그러나 이렇게 진화적으로 보존된 상호작용에 의거한 단백질간의 상호작용의 예측은 실제로 단백질 신약의 대부분을 차지하는 항체와 항원간의 상호작용을 효과적으로 예측하기는 어려웠다. 특정한 단백질을 인식하는 항체는 인식하는 단백질에 따라 고유한 서열을 가지고 있고, 세포 내에 존재하는 상호작용하는 단백질처럼 진화적인 정보를 이용하기는 어렵기 때문이다.
결론적으로 단백질 기반의 의약품의 개발에 구조예측이 좀 더 활발하게 사용되기 위해서는 단백질 간의 상호작용의 예측, 특히 항체와 같은 단백질과 항원과의 결합의 예측이 실험으로 얻는 복합체 구조에 비견될 만큼 개선되어야 할 필요가 있다.
PROTAC을 위한 단백질 모델링
요즘 한창 많은 연구개발이 진행되고 있는 분야 중의 하나가 약물 표적이 되는 단백질을 화학물질이나 항체 등으로 저해하는 것이 아니라 세포 내의 단백질 분해기작인 유비퀴틴-프로테오솜 기전에 의해서 분해되도록 유도하는 ‘표적 단백질 분해(Targeted Protein Degradation)'다.[5] 이 중의 대표적인 것이 분해를 목적하려는 표적 단백질을 단백질의 유비퀴틴화를 촉매하는 효소인 유비퀴틴 라이게이즈와 결합하여 복합체를 형성함으로써, 표적 단백질에 유비퀴틴화를 유도하여 단백질 분해를 유도하는 PROTAC(Proteolysis-targeting chimera)이라는 화학물질이다.
PROTAC의 경우 일반적으로 표적 단백질에 결합하는 화학잔기와 유비퀴틴 라이게이즈에 결합되는 부분이 결합된 화학물질로, 정상적인 경우에는 서로 결합할 일이 없는 표적 단백질과 유비퀴틴 라이게이즈간의 결합을 유도함으로써 표적 단백질에 유비퀴틴을 붙여 단백질 분해를 유도하게 된다.
그렇다면 PROTAC 개발에 어떻게 단백질 구조 모델링이 관여할까? PROTAC 연구 초창기에는, 그저 표적 단백질과 유비퀴틴 라이게이즈에 동시에 결합할 수 있는 화학물질을 이용하여 표적 단백질을 유비퀴틴 라이게이즈에 근접시키는 것만으로 단백질 분해를 유도할 수 있다고 생각하였다. 그러나 여러 시행착오를 겪으면서 알게 된 것은, 일단 PROTAC에 의해서 결합이 유도되는 표적 단백질과 유비퀴틴 라이게이즈도 어느 정도는 안정된 복합체 구조를 형성해야 한다는 것이었고, 따라서 표적 단백질과 유비퀴틴 라이게이즈가 어떻게 복합체를 형성하는지에 대한 지식이 PROTAC을 디자인하는데 필수적이라는 것이었다. 효과적인 PROTAC을 디자인하기 위해서는 표적 단백질과 유비퀴틴 라이게이즈에 결합하는 부위 이외에도 이들을 잇는 링커가 중요하며,어떻게 단백질 복합체+PROTAC의 3차 복합체가 형성되는지를 모델링할 필요가 있다.
이를 위해서는 단백질간 상호작용, 특히 정상적인 경우에는 제대로 결합하지 않을 두 개의 단백질이 결합하는 것을 모델링하고, 여기에 화합물이 결합하는 것을 모델링할 필요가 있다. 그러나 앞에서 이야기한 것처럼 단백질 상호작용의 예측, 특히 PROTAC과 같은 화합물이 존재해야만 결합 가능한 단백질들의 복합체를 정확히 모델링하는 것은 아직까지 쉬운 일이 아니다. 몇 가지 시도들이 진행되고 있으나, 현재까지 매우 높은 정확도로 PROTAC에 의한 복합체 형성을 모델링할 수 있는 방법은 존재하지 않는 실정이다. 이렇게 아직 신약개발을 위한 단백질 구조 관련 예측 관련 분야는 앞으로 발전할 여지가 많이 존재하고 있다. 기존에 많이 사용되던 물리화학적인 원리에 기반한 방법에 추가하여 인공지능 기반의 방법들의 발전은 앞으로 발전이 기대되는 분야 중의 하나이다.
단백질 디자인
특정한 단백질에 결합하는 항체를 선별하는 것은 파지 디스플레이(Phage Display)와 같은 기술이 발전에 의해서 주로 실험적인 방법으로 진행되어 왔다. 그렇다면 항체가 아닌 특정한 단백질에 결합하여 어떤 생리적 활성을 가지거나 아니면 기존의 생리적 활성을 억제할 수 있는 새로운 단백질을 만들 수는 없을까?
이러한 연구는 단백질 디자인(Protein Design) 연구와 깊이 관련되어 있다. 단백질 디자인은 학술적으로 생각한다면 단백질의 입체구조를 서열로부터 예측하는 구조예측 문제의 역함수적인 문제이다. 구조예측이 이미 알고 있는 서열이 가장 안정된 에너지를 가지는 구조를 찾는 문제라면, 단백질 디자인은 정해진 구조에서 가장 안정된 에너지를 가지도록 구조를 이루는 서열을 찾는 문제가 된다. 단백질 디자인의 초창기에는 자연계에서 발견된 단백질 구조에 맞는 최적의 서열을 찾는 문제(고정 골격 디자인 Fixed-backbone design)에 대한 연구가 활발했으나, 이후에는 여러가지 2차 구조 요소들을 조합하여 자연계에는 존재하지 않는 새로운 단백질 폴드를 형성하는 서열을 찾는 연구(드 노보 디자인 De novo design)에 대한 연구가 활발히 진행되었다.[6]
알파폴드 등장 이전 단백질 구조예측 연구를 주도했던 워싱턴 대학의 데이비드 베이커 연구그룹에서는 그들의 단백질 예측 파이프라인 '로제타(Rosetta)'를 이용하여 단백질 디자인 분야에서도 선두주자로 군림했다. 사실 하나의 서열에서 다양한 단백질 구조를 형성하고, 이 중에서 가장 에너지적으로 안정적인 구조를 찾는 문제와, 특정한 구조를 가장 효율적으로 형성하는 서열을 찾는 문제는 결국 많은 갯수의 단백질 구조를 예측하고 그 에너지 상태를 계산하는 것으로 종착되므로, 계산의 관점에서는 완전히 동일한 문제인 셈이다. 이들은 워싱턴 대학에 ‘단백질 디자인 연구소(Institute of Protein Design)'를 설립하여, 기존에 자연계에 존재하지 않는 새로운 구조를 가진 많은 단백질을 디자인할 수 있다는 것을 보여주었다. 그렇다면 이렇게 ‘디자인된 단백질’은 신약 창출의 측면에서 어떤 응용이 가능할까? 한 가지 예는 백신의 제작이다. 이들은 2016년, 인공적으로 디자인된 두 종류의 단백질을 이용하여 약 30나노미터(nm)의 직경을 가지는 20면체로 된 ‘단백질 나노입자’를 만들 수 있다는 것을 보였다.[7] 이러한 단백질 나노입자에 인플루엔자 바이러스의 헤마글루틴과 같이 면역원성을 주는 단백질을 연결시키면, 마치 바이러스와 비슷하게 바이러스의 단백질이 결합된 단백질 나노입자를 형성하게 되고, 이러한 나노입자는 생체 내에서 효율적으로 중화항체를 만들게 된다. 2021년, 4종의 인플루엔자 헤마글루틴으로 구성된 나노파티클 백신은 바이러스 변종에 상관없이 이를 중화할 수 있는 강력한 중화항체를 만들어낼 수 있다는 연구가 출간되었다.[8]
또 다른 예로는 자연계에 존재하는 면역신호 조절 단백질인 사이토카인(Cytokine)인 인터루킨-2 (IL-2)의 유사체를 만들어 면역 항암치료에 사용하는 예다.[9] 인터루킨-2는 결합하는 수용체의 종류에 따라서 그 반응이 달라지는데, 인터루킨-2 수용체 알파, 베타, 감마로 이루어진 수용체와 결합하여 일어나는 면역반응과 베타-감마로 이루어진 수용체와의 반응은 서로 다르며, 알파/베타/감마로 이루어진 수용체와 인터루킨-2와의 상호작용이 과도한 면역반응을 유발하여 독성을 일으키는 것으로 알려져 있다. 이들은 인터루킨 베타/감마 수용체와는 결합을 하지만 알파와는 결합하지 않는 인공 단백질을 디자인하였고, 이렇게 만들어진 인공 단백질이 쥐에서 항암 작용을 일으킨다는 것을 보였다. 이러한 예는 자연계에 존재하지 않는 새로운 단백질이 장차 의약품으로 사용될 수 있다는 가능성을 제시해주고 있다.
그렇다면 이러한 단백질 디자인이 의약품으로 사용되기 위해서 넘어야 할 한계는 어떤 것이 있을까? 일단 대부분의 디자인된 단백질 중에서 계산에 의해서 디자인된 단백질이 바로 생물학적으로 유의미한 결합활성을 가지는 경우는 그리 많지 않으며, 돌연변이 유도 및 라이브러리 구축 및 선별 과정을 거쳐서 실험적으로 결합력을 최적화하는 선별 과정을 거친 이후에야 생물학적으로 유의미한 수준의 결합력을 얻을 수 있었다는 것을 주의할 필요가 있다. 즉 인실리코에서 진행되는 ‘이성적 디자인(Rational Design)'에 더해 실제 진화과정과 흡사한 무작위 돌연변이 및 선별 과정을 거쳐야만 의미있는 수준의 활성을 가진 인공 단백질이 만들어진다는 것을 주목할 필요가 있다. 그리고 인체 내에 존재하지 않는 인공적으로 디자인된 단백질은 궁극적으로 면역계에서 외부의 침입자로 인지되어, 항체 및 세포면역 반응 등이 유도되는 면역원성(Immunogenecity)을 보일 수 있고, 이러한 경우는 단백질 디자인에 의해서 만들어지는 인공 단백질의 효과를 감소시키므로, 치료용 단백질을 디자인할 때는 면역 원성을 최소화하는 것 역시 중요한 고려대상이 될 것이다.
이렇게 단백질 디자인에 의한 인공 단백질의 경우 학계의 연구실 차원에서 활발한 연구가 진행되고 있으나, 아직까지 의약품으로 상용화된 사례는 없다. 그러나 단백질 디자인의 효율이 점점 좋아지고 있고, 특히 알파폴드2와 같이 딥러닝의 도입에 의해서 단백질 구조예측의 정확도가 비약적으로 상승한 것을 감안하면, 단백질 구조예측의 성능과 직접적인 관계가 있는 단백질 디자인 역시 조만간 비약적인 발전이 있을 것으로 생각된다.
결론적으로 알파폴드2의 등장에 의해서 단백질 구조예측 분야의 비약적인 발전이 온 것은 사실이지만, 이러한 발전이 실제 인류의 건강과 복지에 직접적인 영향을 주는 새로운 의약품 등 응용 분야로 이어지기까지는 앞으로도 많은 추가적인 연구가 필요할 것으로 보인다. 단백질 구조의 정확한 예측이 가능하게 된 것은 분명히 구조생물학에 있어서 하나의 큰 이정표이지만, 타깃 단백질 구조의 규명이 현대 신약개발의 속도를 결정하는 보틀넥은 아니기 때문이다.
이것으로 7회에 걸친 연재를 통하여 알파폴드2가 탄생하기까지 단백질 구조예측 과정의 진보를 알아보았다. 그러나 알파폴드2에 의한 정확한 예측이 가능하게 된 기반은 지금까지 수만 종류의 단백질의 구조가 실험적인 방법에 의해서 규명되었고, 이러한 기반 위에 알파폴드2의 예측이 가능해졌다. 다음 연재로는 실험적인 방법에 의한 단백질 구조의 역사와 이러한 구조결정이 어떻게 신약개발 등의 분야에 필수불가결한 요소로 자리잡는 과정에 대해서 알아보도록 한다.
참고문헌
1. Mullard, A. (2021). What does AlphaFold mean for drug discovery?. Nature reviews. Drug discovery.
2. Schindler, T., Bornmann, W., Pellicena, P., Miller, W. T., Clarkson, B., & Kuriyan, J. (2000). Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science, 289(5486), 1938-1942.
3. Lensink, M. F., Brysbaert, G., Mauri, T., Nadzirin, N., Velankar, S., Chaleil, R. A., ... & Wodak, S. J. (2021). Prediction of protein assemblies, the next frontier: The CASP14‐CAPRI experiment. Proteins: Structure, Function, and Bioinformatics, 89(12), 1800-1823.
4. Evans, R., O'Neill, M., Pritzel, A., Antropova, N., Senior, A. W., Green, T., ... & Hassabis, D. (2021). Protein complex prediction with AlphaFold-Multimer. BioRxiv.
5. Békés, M., Langley, D. R., & Crews, C. M. (2022). PROTAC targeted protein degraders: the past is prologue. Nature Reviews Drug Discovery, 1-20.
6. Pan, X., & Kortemme, T. (2021). Recent advances in de novo protein design: principles, methods, and applications. Journal of Biological Chemistry, 296.
7. Bale, J. B., Gonen, S., Liu, Y., Sheffler, W., Ellis, D., Thomas, C., ... & Baker, D. (2016). Accurate design of megadalton-scale two-component icosahedral protein complexes. Science, 353(6297), 389-394.
8. Boyoglu-Barnum, S., Ellis, D., Gillespie, R. A., Hutchinson, G. B., Park, Y. J., Moin, S. M., ... & Kanekiyo, M. (2021). Quadrivalent influenza nanoparticle vaccines induce broad protection. Nature, 592(7855), 623-628.
9. Silva, D. A., Yu, S., Ulge, U. Y., Spangler, J. B., Jude, K. M., Labão-Almeida, C., ... & Baker, D. (2019). De novo design of potent and selective mimics of IL-2 and IL-15. Nature, 565(7738), 186-191.







