기사본문
[남궁석의 신약연구史]단백질, '구조생물학'의 시작
입력 2022-04-28 11:05 수정 2022-04-29 09:48
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
켄드류와 퍼루츠가 마이오글로빈과 헤모글로빈의 구조를 X선 결정학으로 최초로 규명한 이후, 과학자들은 다른 단백질의 구조규명에 나섰다. 결국 생명체를 이루는 생체고분자 중에서 가장 중요한 역할을 하는 단백질의 기능을 알기 위해서는 단백질의 구조를 아는 것이 우선이다. 단순히 단백질의 구조를 아는 것을 떠나서 단백질의 구조를 통하여 생물학의 분자적 기전을 파악하는 ‘구조생물학'이 비로소 시작된 것이다.
이러한 구조생물학의 여명이 시작된 1960년대 후반에 많은 사람들이 우선적으로 구조결정을 시도한 단백질은 세포 내에서 화학반응을 촉매하는 효소(Enzyme)의 구조였다.
효소의 구조가 알려준 효소의 작용기작
사실 효소가 단백질로 이루어져 있다는 것은 20세기 초에 이미 알려져 있었지만 효소가 화학반응의 촉매로서 작동하게 되는 본질에 대해서는 의견이 분분했다. 일부 과학자, 특히 당시 생화학계의 주류였던 독일의 학계에서는 효소가 단백질로 이루어져 있지만, 효소의 촉매작용은 단백질이 아닌 단백질에 결합되어 있는 소분자 화학물질인 보결분자단(Prosthetic Group)에 의해서 이루어지고, 단백질은 단지 이러한 보결분자단이 결합되어있는 담체(Carrier)로만 역할한다는 주장이 대세였고, 단백질이 촉매역할을 직접 수행한다는 의견은 상대적으로 소수의견이었다.
그러나 지난회의 연재에서 설명한 섬너의 요소분해효소 결정화는 단백질만으로 촉매작용을 수행한다는 것을 암시하는 결과였다. 이렇다할 보결분자단이 발견되지 않은 요소분해효소 결정을 다시 녹여도 정상적으로 효소활성을 유지하고 있었고, 결정은 순수한 단백질 분자로만 구성되어 있으므로 단백질만으로 효소의 촉매기능이 작동한다는 것을 의미한다. 그렇다면 단백질이 어떻게 화학적 촉매반응을 수행할까? 이의 정확한 화학적인 기작을 알기 위해서는 단백질을 구성하는 아미노산들이 단백질 내에서 어떻게 배열되어 화학반응에 참여하는 물질과 상호작용하는지를 알아야 하며, 결국 단백질의 원자수준에서의 3차구조를 알아야 한다.
가장 먼저 3차원 구조가 규명된 효소는 라이소자임(Lysozyme)이다. 라이소자임은 계란의 흰자, 눈물 등 동물의 다양한 체액에 들어 있으며, 세균의 세포벽을 녹여 항균작용을 나타내는 효소다. 1965년 데이비드 필립스(David C. Phillips)가 이끄는 연구팀은 계란 흰자 유래의 라이소자임을 결정화하고 구조를 규명했다.[1] 사실 라이소자임이 구조가 규명된 최초의 효소가 된 것은 그 생물학적인 중요성보다는 단백질 입수의 용이함(계란 흰자와 같은 쉽게 얻을 수 있는 재료에서 다량으로 정제 가능하다), 그리고 라이소자임은 다른 단백질에 비해서 유독 결정이 잘 만들어지고, 만들어진 결정은 X선에서 잘 회절하기 때문이었다.
이렇게 얻어진 라이소자임의 구조는 기존에 밝혀진 단백질 구조인 마이오글로빈과 헤모글로빈이 알파나선(alpha-helix)으로만 구성되어 있는 것에 반하여 알파나선과 베타쉬트(beta-sheet)가 섞여 있는 최초의 단백질 구조였다. 그렇다면 이러한 라이소자임의 구조는 라이소자임이 효소로서 작용하는 것에 대한 어떤 정보를 주게 되었을까?
라이소자임은 세균의 세포벽의 구성분인 펩타이도글리칸(Peptidoglycan)을 구성하는 단위체인 N-아세틸뮤람산(N-acetylmuramic acid, NAM)와 N-아세틸-D-글루코사민(N-acetyl-D-glucosame) 사이의 결합을 끊는 효소이다. 다당류의 결합을 끊기 위해서는 일단 다당체 분자가 라이소자임에 결합해야 하며, 라이소자임의 구조의 가운데에는 긴 다당류가 붙을 수 있는 움푹한 영역이 존재하고 있었다. 그리고 실제로 가수분해 반응을 매개하는 아미노산이 존재해야 한다. 필립스 연구팀에서는 라이소자임의 입체구조에 기반하여 다당류가 붙을 수 있는 영역에 마주보고 있는 2개의 산성 아미노산, 즉 52번째 아스파르트산과 35번째 글루탐산이 촉매작용에 참여하는 효소반응 기전을 제시하였다. 52번째 아스파르트산이 NAM과 NAG을 연결해 주는 글리코시딕 결합(Glycosidic bond)에 결합하여 중간체를 형성하고, 나중에 물 분자가 들어와 가수분해 반응이 일어나며 형성되는 양성자를 35번째 글루탐산이 가져가는 메커니즘이다. 이러한 메커니즘은 이후의 구조생물학 및 생화학적 연구를 통하여 검증되었다.

▲그림 1. 라이소자임(Lysozyme)의 반응과 단백질 구조. 라이소자임은 세균 세포벽을 형성하는 물질인 펩타이도글리칸의 단위체인 NAM(N-acetylmuramic acid)과 NAG(N-acetyl-D-glucosame) 사이의 결합을 끊는 효소이다. 라이소자임 구조에서 분해되는 다당류는 라이소자임의 가운데에 파인 홈에 결합되어 있으며, 라이소자임에 있는 두 개의 산성 아미노산인 35번째 글루탐산(Glu35)과 52번째 아스파르트산(Asp52)가 촉매 작용에서 중요한 역할을 한다.
라이소자임 다음으로 밝혀진 효소의 구조들은 단백질 분해효소인 키모트립신과 트립신이었다. 1967년 처음으로 구조가 밝혀진 단백질 분해효소인 키모트립신은 효소의 촉매반응을 수행하는 활성자리에 세린이 존재하는 세린 프로테이즈(Serine Proease)로 불리는 계열의 단백질 분해효소다.[2]
키모트립신에서 촉매반응의 핵심을 담당하는 세린은 195번째 아미노산이다. 그러나 세린이 단백질 분해에서 역할하기 위해서는 다른 아미노산이 필요한데, 57번째 아미노산인 히스티딘과 102번째 아미노산인 아스파르트산이며, 이들을 합쳐서 '촉매반응 3인방(Catalytic Triad)'이라고 부른다. 195번 세린이 단백질의 펩타이드 결합과 직접 반응하여 중간체를 형성하기 위해서는 세린이 가지고 있는 양성자(H) 분자를 다른 아미노산에 넘겨주어고 이온화되어야 하는데, 여기서 이 역할을 하는 것이 양성자를 붙였다 뗐다 할 수 있는 능력을 가지는 히스티딘이다. 57번째 히스티딘은 세린으로부터 양성자를 받고, 히스티딘은 102번째 아스파르트산과의 이온결합에 의해서 안정화된다.
이러한 반응기전은 아직 키모트립신의 구조가 알려지지 않았을 때부터 생화학적 실험결과에 기반하여 가설로 제시되었으나, 실제로 이러한 아미노산의 상호작용이 효소의 활성에 영향을 미치는지는 확인되지 않고 있었다. 그러나 일단 키모트립신의 구조가 규명되고, 세린, 히스티딘, 아스파르트산의 ‘촉매반응 3인방' 아미노산들이 키모트립신의 중심부에 모여 있고, 이들이 실제로 양성자를 주고받을 수 있을 만큼 근접한 거리에 있다는 것이 확인된 이후에야 이러한 반응기전이 실제로 존재함이 입증되게 되었다. 즉, 생체 내에서 화학반응을 일으키는 주 원동력인 효소의 촉매 기능의 본질 역시 단백질 구조의 규명으로써 비로소 완벽히 규명된 셈이다.
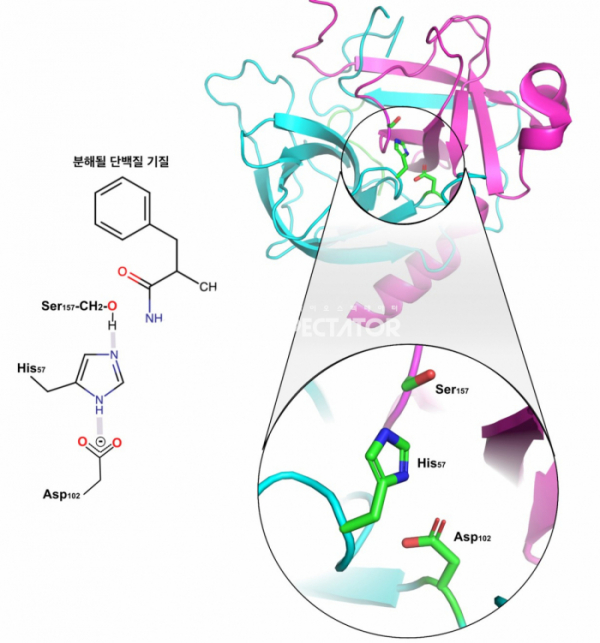
▲그림 2. 단백질 분해효소인 키모트립신(Chymotrypsin)의 활성 메커니즘과 단백질 구조. 키모트립신은 세린 프로테이즈(Serine Protease)라고 불리는 계열의 단백질 분해효소로써, 단백질의 펩타이드 결합을 끊어내는 데는 세린, 히스티딘, 아스파르트산의 3개의 아미노산이 필요하다. 세린(Ser157)이 이온화되어 펩타이드 결합을 공격하며, 이를 위해서 세린의 양성자는 히스티딘 (His57)이 가져가며, 아스파르트산(Asp102)이 히스티딘과 이온 결합을 하여 안정화시키는 구조다. 키모트립신의 단백질 구조에서 세린, 히스티딘, 아스파르트산은 모두 이온 결합이 가능한 약 2 옹스트롬 내의 위치에 일렬로 배열되어 있다.
세포 골격 단백질의 구조
단백질 결정학이 등장한 이후 주로 효소를 중심으로 단백질 구조가 규명되기 시작하였지만, 1970년대 이후부터는 효소와 같은 촉매반응을 하는 단백질 이외에도 세포 내에서 다양한 역할을 하는 류의 단백질로 관심이 옮겨갔다.
이러한 단백질 중의 하나는 세포골격의 중심 역할을 하는 단백질인 액틴(actin)이었다. 근육의 수축, 세포 분열, 세포의 이동 등 수많은 생명현상에 관여하는 단백질인 액틴은 세포 내에서는 단량체 형태인 G-액틴(Globular actin)과 액틴 단량체가 모여서 형성된 필라멘트 형태인 F-액틴(Filament actin)의 두 가지 형태로 존재한다. 그렇다면 이러한 액틴의 구조는 어떻게 얻을 수 있었을까?
일단 수많은 G-액틴이 결합하여 형성되는 필라멘트 형태의 F-액틴의 경우 결정을 얻는 것은 불가능했다. 단백질 결정을 얻기 위해서는 결정을 형성하는 단백질의 조성이 일정해야 하는데, F-액틴의 경우 수십개의 G-액틴으로부터 형성된 짧은 필라멘트부터 수천개의 액틴이 중합되어 만들어진 매우 긴 필라멘트까지 다양한 조성을 가지고 있다. 결정이 되기 위해서는 결정의 구성물이 되는 단백질이 동일한 화학조성을 가지고 있어야 하는데, 형성된 F-액틴은 각각 서로 다른 중합체를 가지고 있으므로, 결정화가 불가능하였다.[3]
그리고 G-액틴의 결정을 얻는 것도 쉬운 일이 아니었다. 단백질의 결정이 형성되기 위해서는 높은 농도의 단백질이 필요하며, 이러한 단백질이 침전과 수용액 상태로 존재하는 중간상태에서 결정화가 이루어진다. 그러나 세포 내에서 분리한 액틴을 결정을 형성할 수 있을 정도의 높은 농도로 농축시키면 G-액틴은 F-액틴으로 중합되어 버리고, 따라서 결정은 형성되지 않게 된다. 따라서 G-액틴의 결정을 얻기 위해서는 액틴의 중합을 억제시켜 높은 농도로 액틴을 농축해도 F-액틴으로 중합되지 않는 조건이 필요했다. 그렇다면 어떻게 하면 액틴의 중합을 억제할 수 있을까? 이러던 와중에 우연히 연구자들은 G-액틴은 DNA 가수분해효소 I(DNase I)과 매우 강하게 결합하며, DNA 가수분해효소와 결합한 G-액틴은 결정을 형성할 수 있을 정도로 높은 농도로 농축되면서도 F-액틴으로의 중합이 억제된다는 것을 발견했다. 이러한 성질을 이용하여 DNA 가수분해효소와 결합된 G-액틴의 구조가 1990년 규명되었다.[4] 그렇다면 G-액틴이 모여서 형성되는 F-액틴은 어떻게 구조를 알 수 있었을까?
이를 이해하기 위해서 1953년 제임스 왓슨, 프랜시스 크릭이 제창한 DNA 이중나선의 구조모델을 만들 때 매우 중요한 정보를 제공했던 로절린드 프랭클린(Rosalind Franklin, 1920-1958)의 DNA X선 회절 사진에 대한 이야기를 할 필요가 있다. 당시 로절린드 프랭클린의 DNA 샘플 역시 단일 결정을 얻지는 못하였으며(당시에는 합성 DNA 기술이 없었으므로 크기가 일정한 DNA를 얻을 수 있는 방법이 없었다) 그가 분석에 사용된 것은 필라멘트화된 DNA 다발이었다. 이러한 비결정 물질의 경우에도 X선에 노출시키면 어느 정도의 회절을 보여주며, 원자수준의 구조 정보를 담고 있지는 않지만, DNA가 이중나선이라는 정도의 샘플 구조에 대한 정보를 얻을 수 있게 된다.[5] (당시에 왓슨, 크릭은 프랭클린의 이러한 회절 데이터에 착안하여 DNA의 이중나선 구조 모델을 만들었지만, 이 데이터는 DNA의 단일 결정으로부터 나온 회절 데이터가 아닌 관계로 원자 수준의 DNA 구조에 대한 정보는 들어 있지 않았다. 실제로 이중나선 DNA의 결정이 만들어지고 원자 수준의 DNA 모델이 만들어져서 왓슨-크릭의 모델이 확실히 검증된 것은 1970년대 말, 즉 왓슨/크릭이 이중나선 모델을 발표한 후 거의 30년이 넘어서였다) 이러한 실험방법을 섬유회절(Fiber Diffraction)이라고 한다.
1958년 암으로 36세의 젊은 나이로 숨진 로절린드 프랭클린은 죽기 전 케네즈 홀메즈(Kenneth Holmes)라는 대학원생을 지도하였는데, 그는 이후 프랭클린이 DNA 구조에 대한 정보를 얻어낼 때 사용한 섬유회절 방법이 액틴 필라멘트에서도 적용가능하다는 것을 알아냈다. 결정화가 불가능한 필라멘트 형태의 F-액틴의 경우에도 X선에 노출시키면 어느 정도의 회절을 보여주며, 원자 수준의 구조정보를 담고 있지는 않지만, 전체적인 필라멘트의 구조를 암시하는 정보를 얻을 수 있었다. 이 정보로 유추한 액틴 필라멘트의 구조는 2개의 액틴 분자가 맞닿아서 이어지는 나선 형태를 하고 있었으며, 단백질 결정을 통해서 얻은 G-액틴의 구조를 기본 단위체로 하여 섬유회절로 얻은 필라멘트 구조에 맞추어 구성한 액틴 필라멘트의 모델이 1990년 등장했다.[6]
이렇게 세포 골격의 중심이 되는 액틴 필라멘트의 구조가 등장한 후, 액틴과 상호작용하여 근육수축 및 물질이동을 일으키는 모터 단백질인 마이오신(Myosin), 그리고 액틴과 더불어 세포 골격을 이루는 또 다른 주요 단백질인 튜블린(Tubulin), 그리고 튜블린과 상호작용하는 모터 단백질인 키네신(Kinesin)과 다이네인(Dynain) 등 ‘세포의 부품' 역할을 하는 수많은 단백질의 구조가 1990년대 이후부터 속속 등장하게 되었다.
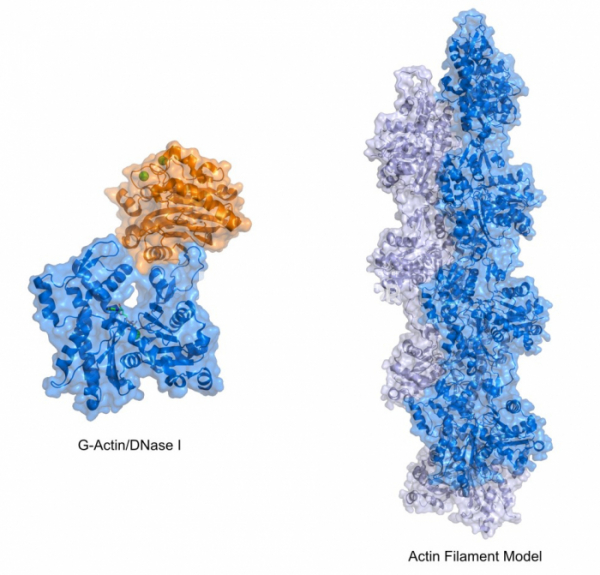
▲그림 3. 액틴 필라멘트의 단위체인 G-액틴(좌) 와 이를 기반으로 구축된 액틴 필라멘트 모델(우). 우리 몸에서 근육 수축, 세포 이동 등 수많은 일에 관여하는 세포골격(Cytoskeleton)의 중심 구성물인 액틴의 구조는 1990년에 이르러서 규명되었다. G-액틴의 구조는 필라멘트로의 중합을 억제하기 위하여 DNA 가수분해효소 I(DNase I, 왼쪽 그림의 주황색으로 표시된 부분)와의 복합체로 결정화되었고, 결정화가 불가능한 액틴 필라멘트는 섬유회절법(Fiber Diffraction)이라는 엑스선 회절 기법을 통하여 얻어진 필라멘트의 정보에 기반하여 G-액틴의 구조로부터 모델링되었다.
재조합 단백질 기술과 유전체 기술과 구조생물학과의 관계
이렇게 초창기의 단백질 구조는 주로 손쉽게 다량의 단백질을 얻을 수 있는 효소의 구조를 중심으로 규명되었다. 당연히 손쉽게 다량의 단백질을 얻을 수 있는 단백질은 수가 한정되어 있고, 결정화되는 단백질 역시 한정되어 있으므로 단백질 구조가 풀리는 속도는 매우 느렸다.
1971년, 그동안 규명된 단백질 구조에 대한 정보를 취합한다는 목적으로 미국 브룩헤이븐 국립연구소의 월터 해밀턴(Walter Hamilton)은 단백질 데이터 뱅크(Protein Data Bank, PDB)를 설립하였다(PDB 가 설립된 것은 DNA의 유전정보를 저장하는 데이터베이스인 진뱅크 GenBank보다도 거의 10년 이전의 일이다). 그런데 그때까지 3차원구조가 풀린 단백질은 10종 미만이었고, PDB에 처음으로 등록된 단백질은 모두 7종에 불과하였다. 반면 1977년 프레데릭 생거에 의해서 DNA의 염기서열을 결정하는 방법이 개발된 이후, 유전자의 DNA의 서열을 통하여 단백질의 아미노산 서열을 손쉽게 얻을 수 있게 되었으므로, 곧 우리가 알고있는 단백질의 아미노산 서열의 숫자는 급증하게 되었다. 그러나 단백질의 구조가 규명되는 속도는 현저하게 느렸다. 특히 단백질 구조를 얻기 위하여 결정을 얻기 위해서는 다량으로 순수한 단백질을 얻을 수 있어야 하는데, 단백질의 종류가 많고, 다량으로 순수한 단백질을 얻기 어려운 고등생물 유래의 단백질의 구조를 규명하는 것은 불가능에 가까웠다.
그러나 1980년대 이후 유전자조작에 의해서 단백질의 유전자를 확보하고, 이를 대장균 등에 넣어서 많은 양의 단백질을 생산하는 재조합 단백질 기술이 등장하면서 이것이 단백질 구조결정에도 큰 영향을 주기 시작하였다. 일단 천연 조직 유래에서 다량의 단백질을 얻기 힘들었던 단백질도 유전자만 확보되어 있다면 재조합 단백질 기술로 다량의 단백질을 얻을 수 있게 되었기 때문에 이를 이용하여 결정화를 시도할 수 있게 되었고, 따라서 기존에는 얻지 못하던 수많은 단백질의 구조를 얻게 되었다.
그리고 진핵생물의 경우 여러개의 도메인(Domain)으로 구성되어 있는 단백질이 많은데, 이러한 도메인들은 뚜렷한 3차원구조를 가지고 있지만, 이 도메인들은 뚜렷한 형태를 가지고 있지않은 무정형의 아미노산 서열로 연결되어 있는 경우도 있고, 이러한 경우 전체 단백질로써는 결정화하기 어려운 경우도 많았다. 이러한 경우에도 단백질의 도메인에 해당하는 유전자 부분을 따로 떼어 재조합 단백질을 만들어서 결정화하고 구조를 결정할 수 있다는 것을 알게 되었다.
이렇게 분자생물학/재조합 단백질 기술의 발전은 단백질 구조 규명을 직접적으로 촉진시켰다. 결정적으로 1990년대 말부터 생물체의 유전정보가 전부 밝혀져서 본격적으로 ‘유전체 시대'가 개막된 이후 이는 더욱 가속되었다. 단백질의 구조는 단백질 서열에 비해서 훨씬 보존되기 때문에 자신이 직접 연구하는 종의 단백질이 아닌 다른 종의 단백질이라고 하더라도 서열이 유사한 같은 종류의 단백질이라면 구조가 보존된다. 가령 인간 유래의 단백질의 결정화에 실패하여 구조를 얻지 못한다고 하더라도 다른 동물, 혹은 곤충이나 효모, 심지어 세균의 단백질이라고 하더라도 구조가 보존되는 경우가 많이 있고, 이러한 경우 구조 규명이 가능한 어떤 구조라도 규명할 수 있다면 충분히 유용한 정보를 얻을 수 있게 된다. (실제로 켄드류가 규명한 최초의 단백질 구조인 마이오글로빈의 경우에도 켄드류가 규명한 마이오글로빈은 고래의 마이오글로빈이었다) 만약 알려져 있는 유전체의 정보가 많으면 많을수록 다양한 생물종에 보존되어 있는 다양한 단백질의 구조결정을 시도해 볼 수 있기 때문에 구조를 풀 수 있는 가능성이 높아진다. 이렇게 다양하게 축적된 유전체 정보에 근거하여 많은 단백질들이 재조합 단백질 기술로 생산되고, 구조가 결정되었다. 1999년에 이르러 PDB에 기탁된 구조는 1만개를 넘었으며, 2008년에는 5만개, 2014년에는 10만개가 넘었다.[7] 물론 단백질 구조가 축적되는 속도보다 훨씬 빠른 속도로 생물의 DNA 서열이 결정되었고, DNA 서열로부터 유추된 단백질의 서열이 데이터베이스에 축적되는 속도는 단백질 구조가 결정되는 속도에 비해서 훨씬 더 빨랐다. 즉, 단백질의 서열은 알고 있지만 구조를 모르는 단백질은 점점 더 많이 쌓여갔다.
유전체학과 함께 X선 결정학에 의한 단백질 구조결정의 발전을 가속화한 기술은 '방사광 가속기(Synchrotron)'다. 방사광 가속기(Synchrotron)에서는 통상적인 실험실에 있는 X선 발생 장치에서 배출되는 것보다 훨씬 더 강한 X선을 얻을 수 있다.[8] 이러한 강한 X선을 단백질 결정에 쬐면 좀 더 높은 해상도로 회절 데이터를 얻을 수 있고, 회절 데이터를 얻는 시간도 훨씬 단축되었다. 물론 강한 X선을 단백질 결정에 쬐면 강한 방사선 때문에 결정이 빨리 손상된다. 그러나 이러한 문제는 결정을 액체질소 등을 이용하여 극저온으로 얼리면서 데이터를 수집하면서 피할 수 있다는 것을 알게 되었다. 이렇게 방사광 가속기의 이용과 극저온 결정학의 도입으로 단백질 구조의 결정 속도는 급격히 빨라졌다.
켄드류와 퍼루츠가 구조를 풀던 시절에는 단백질 결정에 X선을 쬐어 나온 회절 패턴을 X선 필름에 감광하여 현상하고, 이러한 사진을 며칠에 걸쳐 수백장을 찍고 회절패턴을 구성하는 점들의 위치와 강도를 일일히 측정해야 하는 몇달에서 몇년이 걸리는 작업이었지만, 강력한 방사광 가속기에서 나오는 X선을 회절에 이용하면 불과 수십분 안에 데이터 수집 작업을 마칠 수가 있게 된다. 따라서 단백질 구조를 푸는 과정에서 X선 회절 데이터를 얻는 과정은 더이상 병목지점이 되지 않게 되었다.
이러한 기술적 발전에 의해 순수한 단백질을 얻기 쉽고 결정화가 쉬운 단백질의 구조부터 우선적으로 규명하여 생명현상의 기본 부품으로써의 단백질의 기능을 파악하던 단백질 결정학은 1990년대 말부터 인간의 생리와 질병에 중요한 역할을 하는 단백질의 구조를 규명하는 쪽으로 촛점이 맞춰지게 되었다.
참고문헌
1. Phillips, David C. (1966). The Three-Dimensional Structure of an Enzyme Molecule. , 215(5), 78–90. doi:10.1038/scientificamerican1166-78
2. Matthews, B. W., Sigler, P. B., Henderson, R., & Blow, D. M. (1967). Three-dimensional structure of tosyl-α-chymotrypsin. Nature, 214(5089), 652-656.
3. Dominguez, R., & Holmes, K. C. (2011). Actin structure and function. Annual review of biophysics, 40, 169-186.
4. Kabsch, W., Mannherz, H. G., Suck, D., Pai, E. F., & Holmes, K. C. (1990). Atomic structure of the actin: DNase I complex. Nature, 347(6288), 37-44.
5. Franklin, R. E., & Gosling, R. G. (1953). Molecular configuration in sodium thymonucleate. Nature, 171(4356), 740-741.
6. Holmes, K. C., Popp, D., Gebhard, W., & Kabsch, W. (1990). Atomic model of the actin filament. Nature, 347(6288), 44-49.
7. Abad-Zapatero, C. (2012). Notes of a protein crystallographer: on the high-resolution structure of the PDB growth rate. Acta Crystallographica Section D: Biological Crystallography, 68(5), 613-617.
8. Holmes, K. C., & Rosenbaum, G. (1998). How X‐ray Diffraction with Synchrotron Radiation Got Started. Journal of synchrotron radiation, 5(3), 147-153.