기사본문
[남궁석의 신약연구史]'암과의 전쟁' 선포와 바이러스
입력 2017-10-25 15:14 수정 2017-10-25 15:14
남궁석 충북대 교수
'암과의 전쟁' 선포와 바이러스
자넷 로울리 (Janet D. Rowley) 가 필라델피아 염색체의 정체를 밝히던 1971년은 암 연구의 역사에 있어서 획을 긋는 일대 사건이 일어난 해이다. 1971년 12월 닉슨 대통령은 ‘National Act of Cancer’ 라는 법안을 서명하고 ‘암과의 전쟁’ (War on Cancer)을 선포하였다. 이 계획은 1972년부터 3년간 15억 달러의 연구비를 투자하여 미국 독립 200주년이 되는 1976년까지 암 정복을 위한 가시적인 성과를 내겠다는 야심찬 계획으로 ‘암과의 전쟁’ 을 지휘할 콘트롤 타워로써 미 국립보건원 (NIH) 내에 국립 암 연구소 (NCI, National Cancer Institute) 가 세워졌다. 그렇다면 ‘암과의 전쟁’ 이 선포된 배경은 무엇이었을까?
1945년 프랭클린 루즈벨트 대통령의 과학보좌관인 바네바 부시 (Vannevar Bush)가 백악관에 제출한 보고서인 ‘과학, 끝없는 프론티어' (Science, the Endless Frontier)[1] 는 2차 대전 이후 미국의 연방정부의 과학지원의 기반이 되었다. 그는 이 보고서에서 평화시 과학 연구의 중요한 목표로 ‘질병과의 전쟁’을 들었으며, 이를 위해서는 질병 자체의 이해가 필수적이므로 질병 관련의 생명현상에 대한 폭넓은 연구를 지원해야 한다고 역설했다. 이를 계기로 미 국립보건원 (National Institute of Health) 를 통한 의생명과학 분야에 대한 연구비는 1960년대 중반 10억 달러를 넘을 정도로 급격하게 증대하였다[2]. 그러나 이렇게 증가한 연구비에 비해서, 페니실린처럼 일반 대중들이 체감할 수 있는 의학적인 진보는 미미했고, 실질적으로 질병 치료와 관련된 연구 대신 과학자들의 과학적 호기심을 충족시키기 위한 연구에 납세자의 혈세가 낭비된다는 비판도 점점 커져갔다.

▲그림 1 : 1971년 12월 리처드 닉슨 (Richard Nixon) 당시 미국 대통령에 의해 'National Act of Cancer' 법안이 서명됨으로써 공식적으로 '암과의 전쟁' 이라는 언제 끝날지 알 수 없는 영원한 전쟁이 시작되었다. (출처: https://www.cancer.gov/about-nci/legislative/history/national-cancer-act-1971)
이러한 여론과 함께 때마침 진행되었던 아폴로 계획을 통해 불과 몇 년 만에 인간을 달에 보냈다 무사귀환시키는 상상하기 힘든 대업을 성공한 미국은, 암과 같은 질병에도 아폴로 계획과 유사한 정부 주도의 집중적인 투자가 이어진다면 암 치료에 대한 돌파구를 열 수 있지 않을까라는 생각으로 ‘암과의 전쟁’ 을 적극적으로 추진했다. 물론 생물학자 중에서는 콜롬비아 대학의 암생물학자인 솔 스피겔만(Sol Spigelman)처럼 부정적인 반응을 보인 사람도 있었다. 그는 “현재 벌어지는 전면적인 암과의 전쟁은 마치 뉴턴의 중력법칙을 모르는 상태에서 인간을 달에 착륙시키겠다는 것과 마찬가지이다"라고 언급하였다[3]. 즉, 아폴로 계획에서 인간을 달에 착륙시키는 데 필요한 모든 과학적 지식은 이전에 확립되었고, 인간을 달에 보냈다 귀환시키는 것은 어디까지나 얼마나 이를 경제적이고 효율적으로 수행시키느냐는 공학적 문제인 데 반하여, ‘뉴턴의 중력법칙’ 에 해당하는 암 발생의 근본적인 원리도 파악하지 못하는 채 암 치료의 돌파구를 찾겠다고 한 당시의 계획이 얼마나 무모한 것이었는지를 잘 보여주는 언급이다.
그렇다면 당시에 ‘암을 몇 년 안에 치료할 수 있다’ 라는 지금 생각하면 너무나 낙관적인 계획이 어떻게 입안되었으며, 여기서는 어떤 연구들이 시도되었을까? 이전 연재에 설명한 것처럼 1970년대 초는 암의 주 원인이 유전적인 손상인지, 아니면 바이러스와 같은 병원체인지도 확실히 모르던 시절이었다. 그러나 지난 연재에서 언급된 페이튼 라우스의 RSV 등의 레트로바이러스 (Retrovirus) 가 인간 암의 주 원인이 아닐까 하는 생각이 ‘암과의 전쟁’ 의 주된 원동력이 되었다[4]. 특히 1970년 RNA 바이러스인 레트로바이러스를 DNA 로 바꾸고, 숙주의 DNA 내로 삽입되도록 하는 역전사효소 (Reverse Transcriptase)가 발견된 이래[5], 만약 대개의 암이 레트로바이러스에 의해서 일어나고, 레트로바이러스의 증식에 필수적인 역전사효소 (Reverse Transcriptase) 를 억제하여 바이러스의 증식을 막는다면 페니실린과 같이 많은 종류의 박테리아를 억제하는 ‘기적의 약’ (Miracle Drug)을 얻은 것처럼 암에 대한 ‘기적의 약’ 을 창출할 수 있을 것이라는 막연한 기대를 가지고 많은 연구자들은 암을 유발하는 레트로바이러스를 찾는데 많은 노력을 기울였고 ‘암과의 전쟁’에서 투자되기 시작된 연구비의 상당수도 여기에 투자되었다[6].
그러나 이러한 낙관적인 기대와는 달리 사람에게 암을 일으키는 레트로바이러스는 거의 발견되지 않았다. 사실 애초에 대개의 암은 전염성이 없다는 것은 그 당시에도 알려져 있었다는 것을 생각하면 이러한 기본 가정 자체가 상당히 문제있는 가정이었던 셈이다. 암을 일으키는 레트로바이러스가 발견되지 않았으므로 당연히 기대했던 암을 치료하는 ‘기적의 약’ 이 탄생할 수는 없었다. 그렇다면 1970년대 ‘암과의 전쟁’ 에서 투자된 연구비는 그저 헛되이 낭비된 것이었을까? 꼭 그렇게 생각하기는 힘든 부분이 있다. 1970년대에 레트로바이러스 연구에 쏟았던 노력은 전혀 기대하지 않던 곳에서 결실을 거두게 되기 때문이다. ‘암을 유발하는 레트로바이러스’ 를 찾기 위해서 쌓은 지식은 1980년대초에 갑작스럽게 문제가 되기 시작한 레트로바이러스 유래의 질병, 그것도 인류의 존속을 위협하는 ‘21세기의 흑사병’ 으로 묘사되었던 에이즈(AIDS)의 병원체(Human immunodefiency virus, HIV) 를 파악하고, 그 치료수단을 개발하는데 바로 적용되었다[6]. 그리하여 한때 21세기의 흑사병으로 생각되던 에이즈는 이제 관리가 가능한 만성병이 된 것을 생각하면 ‘암과의 전쟁’ 에서 투자된 노력은 기대하지 않은 곳에서 결실을 거둔 셈이다. 그리고 무엇보다도 1970년대에 시작된 암을 유발하는 바이러스에 대한 연구는 암의 분자적인 근원을 파악하는데 결정적인 역할을 하게 된다. 암을 당장 치료하는 ‘기적의 약’ 의 개발까지는 한참 기다려야 했지만 말이다.
바이러스에 존재하는 ‘암 유전자’ 와 그 유래
기대와는 달리 인간에게서 암을 유발하는 레트로바이러스는 별로 발견되지 않았지만, 적어도 조류세포나 설치류 등의 모델 시스템에서 발견된 RSV등의 암을 유발하는 바이러스는 분명히 암을 유발하는 것은 사실이었다. 그렇다면 어떻게 이 바이러스가 암을 유발할까?
이 의문이 풀리게 된 계기는 1976년 조류세포에서 암을 유발하는 RSV 에서 암을 유발하는 유전자는 정상적인 조류 세포에도 들어있는 거의 동일한 유전자라는 것이 밝혀진 다음이었다[7]. UCSF의 해롤드 바무스 (Harold E Varmus) 와 J.마이클 비숍 (J.Micheal Bishop) 을 포함한 연구진들은 RSV에는 v-src 이라는 유전자가, 암이 아닌 정상 조류세포에는 c-src 이라는 거의 비슷한 유전자가 존재하며, 이 단백질은 단백질의 타이로신 잔기를 인산화시키는 타이로신 인산화효소 (Tyrosine Kinase) 라는 것이 확인되었다[8]. 그렇다면 정상 세포와 거의 동일한 바이러스의 유전자가 어떻게 암을 유발시킬까? 바이러스 유래의 v-src 단백질은 세포내에 있는 c-src 보다 훨씬 높은 단백질 인산화 활성을 가지고 있음이 밝혀졌다. 즉, 바이러스가 가지고 있는 숙주 유래의 타이로신 인산화 효소 유전자는 정상 세포 버전과 거의 비슷하지만, 단 하나의 차이는 바이러스 유래의 유전자는 정상 버전보다 약간 짧다는 것이다.
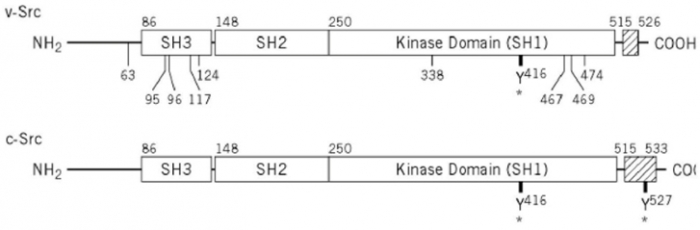
▲그림 2 : 바이러스에 들어있는 v-src 과 정상세포에 들어있는 c-Src 은 정상세포 버전이 C 말단에 조금 더 길다는 매우 미묘한 차이가 있을 뿐이다. 그러나, 이 영역에 존재하는 527번 타이로신 (Y527) 이 인산화되면, 타이로신 인산화효소인 c-Src 의 활성은 자체적으로 조절되게 된다. 반면에 이 부분이 존재하지 않는 v-src 은 ‘브레이크’ 가 풀려 항상 활성을 가지게 된다.
그 후에 정상세포 버전의 c-src의 끝부분 (바이러스에는 존재하지 않는)에 있는 타이로신 잔기(527번째 타이로신 잔기, Y527)에 인산화가 일어나면 타이로신 인산화효소의 활성이 억제된다는 것이 밝혀졌다[9]. 즉 바이러스 버전의 v-src에는 결여되어 있지만 정상 세포 버전에는 있는 부분이 효소의 활성을 억제하는 ‘브레이크’ 처럼 작동하는 것이다. 즉 정상 세포에서는 ‘브레이크’ 에 의해서 작동이 조절되는 타이로신 인산화 효소 유전자가 바이러스가 정상 세포의 유전자를 ‘낚아채는’ 과정에서 약간 변형이 생기게 되고, 이렇게 ‘브레이크’ 가 망가진 타이로신 인산화효소 유전자는 바이러스가 감염될 때 세포 내에서 만들어지고, 이렇게 조절 기능이 망가진 인산화효소가 세포 내에서 항상 작동하면서 암이 유발된다.

▲그림 3: 해롤드 바무스(Harold E Varmus)와 마이클 비숍(Micheal J Bishop) 은 레트로바이러스가 어떻게 암을 유발하는지를 규명하였고, 이들은 1989년 노벨 생리의학상을 수상한다. 이들의 발견은 단지 레트로바이러스의 암 유발을 넘어서 암유전자 (Oncogene) 의 분자적 이해의 기반을 마련한다.
오늘날 이들이 발견한 ‘암을 유발하는 유전자’는 ‘암유전자’ (Oncogene) 라고 부른다. ‘암 유전자’ 는 대개 정상 세포에 존재하고 있는 ‘원암 유전자’ (Proto-oncogene) 이 변형되어 ‘브레이크’ 가 고장난 것임이 확인되었다. 즉, 바무스와 비숍의 발견은 암이 어떻게 발생하는지의 기전 연구에서 하나의 패러다임을 형성하게 된다. 즉, 그들의 발견은 단순히 하나의 바이러스가 어떻게 암을 유발하는지의 원인을 넘어서, 분자적 수준에서 암이 발생하는 과정을 알아보는 시발점이 된 셈이다. 즉, 암을 바이러스가 일으키는가, 아니면 염색체에서의 변형이 암을 일으키는가와 같은 해묵인 논란이 “바이러스가 전달하는 것은 암을 일으키도록 변형된 인간 유래의 유전자이다” 라는 하나의 이야기로 통합되기 시작한 지점이기도 하다.
곧이어 바이러스 유래의 암유전자와 정상 세포내에 존재하는 원암 유전자 발견자가 속속 발견되기 시작하였다. 그 중에서는 하베이 (Harvey) 와 커스텐 (Kirsten)이 각각 발견한 Rat sarcoma virus 유래의 유전자가 있었다. 이는 각각 H-Ras 와 K-Ras 로 명명되었다[10]. 오늘날 암 연구자/의사 중에서 이 유전자들의 이름을 모르는 사람은 없으리라고 생각하지만, 이들의 이름 자체가 해당 암 유전자가 유래된 바이러스와 그 발견자의 이름에서 기인한다는 것을 과연 얼마나 많은 사람들이 기억할까?
BCR/ABL
최초의 바이러스 유래의 ‘암 유전자’ 가 발견된 이후, 아벨슨과 랩스타인이 발견한 바이러스의 경우에도 ABL이라는 암유전자를 가지고 있었으며 이는 인간의 9번 염색체에 위치하고 있음이 1982년에 확인되었다[11].
이들의 발견은 그때까지 염색체 수준에서 암을 유발하는 이상이라고 알려진 유일한 사례였던 필라델피아 염색체와 CML 과 관련되어 연구되기 시작하였다. 이전 연재에서 설명한 것처럼 이미 필라델피아 염색체는 22번 염색체가 9번 염색체로 전좌 (Transposition) 되어 일어난다는 것이 발견된 상태이다. 그렇다면 9번 염색체에 존재하는 ABL 은 이 과정에서 어떤 변화를 겪을까?
1984년, 22번 염색체가 절단되어 9번 염색체와 만나는 영역은 한정된 영역에 집중되어 있다는 것이 알려졌고, 이 영역을 ‘breakpoint clustered region’ (BCR) 라고 명명되었다[12]. 곧 필라델피아 염색체의 형성에 의해 9번에 존재하는 ABL과 22번에 존재하는 BCR 유전자가 합쳐져BCR/ABL 이라는 융합 유전자가 생긴다는 것을 알게 되었다[13]. 그리고 CML 환자에서 BCR/ABL 융합 유전자의 mRNA와 단백질이 존재한다는 것도 곧 알려졌다[14].
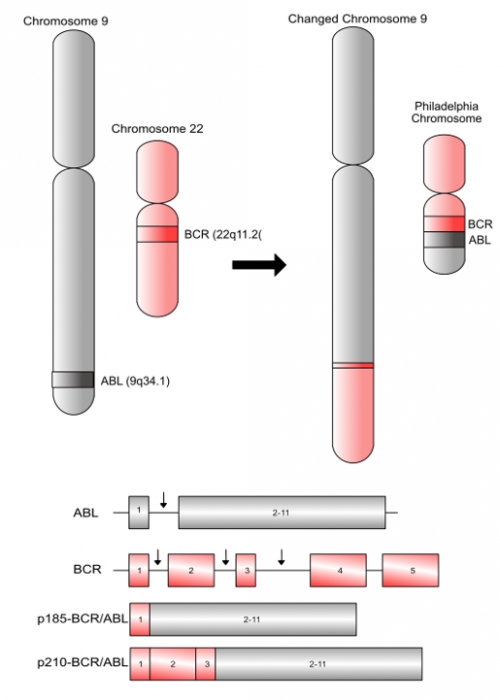
▲그림 4 : 9번 염색체와 22번 염색체가 전좌 (Transposition) 을 일으키면, 9번에 존재하는 ABL 유전자와 22번 염색체에 존재하는 BCR 유전자가 만나 융합 유전자인 BCR/ABL을 형성한다. 22번 염색체의 BCR 유전자의 어떤 부위에서 염색체가 잘리느냐에 따라서 BCR의 1번 엑손 (Exon)과 ABL의 2-11번 엑손이 만난 p185-BCR/ABL 융합 유전자가 형성될 수도 있고, BCR의 1-3번 엑손과 ABL의 2-11번 엑손이 합쳐진 p210-BCR/ABL 유전자가 생길수도 있다.
ABL 역시 암 유전자 src 과 마찬가지로 타이로신 인산화효소의 활성을 가지고 있다는 것을 확인했으며, CML 환자에게서 발생하는 BCR/ABL 은 정상 세포 버전의 ABL 에 비해서 매우 높은 타이로신 인산화효소의 활성을 가짐이 발견되었다[15]. 1990년, 마우스에서 BCR/ABL 융합 단백질 유전자를 발현시키는 것만으로 CML 을 유발시킬 수 있다는 것을 보여줌으로써, BCR/ABL 융합 유전자의 형성이 CML을 유발하는 요인임을 실험적으로 확증하였다[16]
이러한 BCR/ABL의 연구는 이전에 레트로바이러스 유래의 암 유전자와 원암 유전자의 관계, 즉 c-Src 과 v-Src 간의 관계와 유사한 관계가 성립함을 알 수 있다. 즉 세포 내의 신호전달과정에서 매개체로 사용되는 타이로신 인산화 효소가 ‘항상 켜져’ 있도록 변형이 생기면 이것이 암 유전자로 작동한다는 것이다[17]. 즉 여기서 암의 분자적인 기전이라고 볼 수 있는 하나의 패러다임이 출현한다. 즉, 암을 유발하는 유전자 변형은 세포내의 신호전달과정에서 신호를 전달하는 스위치가 ‘항상 켜져 있을때’ 일어난다는 것이다.
오늘날 생물학을 배운 사람이라면 다 아는 이야기지만, 자기 자신의 증식에만 신경쓰면 되는 단세포 생물과는 달리, 다세포 생물에서의 세포는 항상 엄격한 통제 속에서 놓여져 있으며, 세포가 분열해서 증식하는 것과 증식을 멈추는 것은 세포의 외부 환경의 ‘신호’ 를 감지하여 세포내로 전달해주는 신호전달계에 의해서 조절된다. 이러한 엄격한 조절 기전이 붕괴되면 통제받지 않은 세포의 증식이 일어나며 이것은 악성 종양으로 이어진다. 지금 세포생물학 교과서에는 다 나와 있는 이러한 교과서적인 지식이 최초로 정립되는 시대가 바로 1980년대 중반인 셈이다.
가령 BCR/ABL의 경우에는 ABL 유전자 만으로는 정상적인 조건에서는 ‘켜져 있지 않은’ 신호전달 경로가 BCR 유전자와 융합됨으로써 ‘항상 켜져 있도록’ 변형된다. 즉 변형되지 않은 ABL은 N말단의 ‘Cap’ 도메인이 타이로신 인산화 효소를 억제하는 브레이크 역할을 한다. 그러나 BCR/ABL은 이러한 ‘브레이크’ 역할을 하는 부분이 BCR과 융합되며 없어지고, 대신, ‘코일-코일 도메인’ (Coiled-Coil Domain) 이라는 두 개의 단백질을 서로 인위적으로 결합할 수 있게 하는 부분으로 치환된다. 즉 BCR 단백질이 서로 결합하는 성질이 타이로신 인산화효소인 ABL 에 강제로 부여되고, 이렇게 상호결합한 ABL은 서로를 인산화시켜 활성화된다[18].
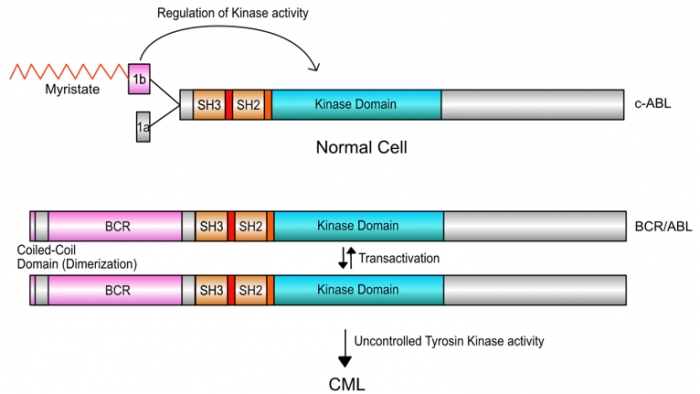
▲그림 5: 정상세포에 있는 변형되지 않은 ABL 단백질은 (c-ABL) 말단에 존재하는Myristate 로 변형된 ‘Cap’ 영역이 부분이 타이로신 인산화효소(Kinase Domain) 부분의 활성을 억제하는 ‘브레이크’ 로 작동한다. 그러나 필라델피아 염색체에 의해서 생성된 BCR/ABL 단백질에는 브레이크로 작동하는 Cap 영역이 없어지고, 대신 여러 개의 단백질 분자를 결합시킬 수 있는 코일-코일 (Coiled Coil) 영역이 BCR 과 함께 딸려온다. 이렇게 변형된 BCR/ABL의 타이로신 인산화효소 (Kinase Domain) 는 서로를 인산화시켜 활성화하고, 조절되지 않은 신호전달을 유발하여 궁극적으로 CML 을 유발시킨다.
물론 CML의 경우에는 매우 특이하게 하나의 유전자의 변형에 의해서 암이 유발되는 사례인 관계로 여러가지 신호전달체계에서의 변형이 복합적으로 관여하는 다른 종류의 암에 비해서 훨씬 더 간단한 ‘고장’ 인 셈이었고, 다른 종류의 암에 있어서는 그렇게 상황은 간단하지 못했지만 말이다.
그러나 이러한 연구가 주는 가장 중요한 의미는 이전에는 미지의 세계로 여겨졌던 암의 발생원인이, 생명현상에 대한 환원주의적 (Reductionist) 접근방식인 분자생물학적 연구에 의해 최초로 암의 원인을 규명할 수 있다는 가능성을 열었다는 것이다. 즉, 특정 단백질 하나 (ABL/BCR) 에 의해서 CML이 유발된다면, 이 단백질의 활성을 억제하는 것으로 특정한 암을 치료하는 방법이 있지 않을까와 같은 희망, 즉 ‘표적항암제’ 에 대한 생각을 학계와 산업계의 연구자들이 가지게 된 시기가 바로 ABL/BCR에 의한 CML의 발생 기전이 어느정도 이해되기 시작한1980년대 말에서 1990년대 초인 셈이다. 물론 암에 대한 이해가 점점 진척되면서 이러한 기대 역시 과다한 기대였다는 것을 깨닫게 되지만, 그것은 한참 뒤의 일이었다.
다음 회에서는 드디어(!) ABL/BCR 을 특이적으로 저해하는 화합물에 대한 이야기를 시작해 보도록 한다.
참고문헌
1. Science, the Endless Frontier, A Report to the President by Vannevar Bush, Director of the Office of Scientific Research and Development, July 1945
2. Dogab Yi. “The Recombinant University:Genetic Engineering and the Emergence of Stanford Biotechnology”, (Chicago: The University of Chicago Press, 2015) p54
3. J.T. Patterson, The Dread Disease:Cancer and Modern American Culture (Cambridge: Harvard University Press, 1989)
4. Huebner, R.J. Todaro, G.J. Oncogenes of RNA Tumor viruses as determinants of cancer, Proc. Natl. Acad. Sci. 1969:64:1087–1094.
5. Mizutani S, Boettiger D, Temin HM. A DNA-dependent DNA polymerase and a DNA endonuclease in virions of Rous sarcoma virus. Nature. 1970:228:424-427.
6. Weinberg R.A., Coming full circle-from endless complexity to simplicity and back again Cell 2014 157:267-271
7. Stehelin, D.; Varmus, H. E.; Bishop, J. M.; Vogt, P. K. (1976-03-11)."DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 260:170–173.
8. Brugge J.S., Erikson R.L. Identification of a transformation-specific antigen induced by an avian sarcoma virus. Nature. 1977;269:346–348; Collett M.S., Brugge J.S., Erikson R.L. Characterization of a normal avian cell protein related to the avian sarcoma virus transforming gene product. Cell. 1978;15:1363–1369; Collett M.S., Erikson R.L. Protein kinase activity associated with the avian sarcoma virus src gene product. Proc. Natl. Acad. Sci. U. S. A. 1978;75:2021–2024; Levinson A.D., Oppermann H., Levintow L., Varmus H.E., Bishop J.M. Evidence that the transforming gene of avian sarcoma virus encodes a protein kinase associated with a phosphoprotein. Cell. 1978;15:561–572.
9. Erpel, T. Courtneidge, S.A. Src family protein tyrosine kinases and cellular signal transduction pathway, Curr Opin Cell Biol. 1995:7:176-182.
10. DeFeo D, et al. Analysis of two divergent rat genomic clones homologous to the transforming gene of Harvey murine sarcoma virus. Proc Natl Acad Sci USA. 1981;78:3328–3332; Ellis RW, DeFeo D, Furth ME, Scolnick EM. Mouse cells contain two distinct ras gene mRNA species that can be translated into a p21 onc protein. Mol Cell Biol. 1982;2:1339–1345;Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci USA. 1982;79:4848–4852.
11. de Klein A., et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765–767; Heisterkamp N., et al. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature. 1983;306:239–242.
12. Groffen J., et al. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99.
13. Shtivelman E., Lifshitz B., Gale R.P., Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315:550–554.
14. Stam K., et al. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N. Engl. J. Med. 1985;313:1429–1433
15. Konopka J.B., Watanabe S.M., Witte O.N. An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell. 1984;37:1035–1042.
16. Daley G.Q., Van Etten R.A., Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–823.
17. Hunter, T. Treatment for chronic myelogenous leukemia: the long road to imatinib. J Clin Invest. 2007 117: 2036–2043.
18. Hantschel O1, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases.Nat Rev Mol Cell Biol. 2004;5(1):33-44.







