기사본문
[남궁석의 신약연구史]인플루엔자 역습&항바이러스제
입력 2019-10-15 14:01 수정 2022-04-21 14:36
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
이전 연재에서 수정란을 이용한 인플루엔자 바이러스의 대량 배양과 이를 불활성화한 백신의 등장으로 인플루엔자에 대한 백신이 등장하였고, 이로 인해 1차 대전 종전 이후 귀국하는 장병들에 의해 전파된 인플루엔자 대유행을 2차 대전 종전 이후에는 막을수 있었다는 에피소드를 소개하였다.
그러나 인플루엔자와 인류와의 싸움은 인플루엔자 백신의 등장으로 종결된 것은 아니었다. 인플루엔자 백신이 등장한 1940년대 이후에도 전세계적으로 인플루엔자 대유행이 있어왔으며, 이로 인해 수많은 사람들이 목숨을 잃었다. 이번 연재에서는 백신의 등장에도 불구하고 계속되는 인플루엔자 대유행의 원인과, 이를 극복하기 위한 새로운 항바이러스 요법의 개발 과정에 대해서 알아보도록 한다.
계속되는 인플루엔자 대유행
1940년대 말 인공배양된 인플루엔자 바이러스를 불활성화시킨 백신이 등장함으로써, 2차 세계대전 종전이후 수백만명의 군인들이 대이동하는 상황에서도 1918년인플루엔자 대유행과 같은 사태는 재현되지 않았다. 그리고 인플루엔자 백신 접종이 일반화됨으로써 백신의 등장 이후 거의 박멸되다시피한 황열병, 폴리오, 천연두와 같이 인플루엔자 역시 이제 더이상 큰 문제가 아닐 것이는 낙관적인 기대가 있었다. 그러나 이러한 기대가 깨지기에는 그리 많은 시간이 걸리지 않았다.
1956년 중국 구이저우성에서 기존에 인간에서 발견된 것과는 전혀 다른 새로운 종류의 인플루엔자 바이러스가 발견되었다. 이 바이러스는 1957년 2월에 싱가포르에서 발견되었으며 4월에는 홍콩에, 그리고 6월에는 미국에서 처음 발병 환자가 발생하였다. 기존의 백신으로 예방되지 않는 새로운 인플루엔자 바이러스에 의해서 미국 내에서만 약 6만9,800명의 사망자가 보고되었다. 다른 인플루엔자 대유행과 마찬가지로 1957년의 아시아에서 유래된 이 인플루엔자의 경우에도 당시에는 정확히 어느정도의 희생을 가져왔는지는 확실하지 않았다. 2016년의 연구는 1957년의 인플루엔자 대유행으로 전세계에서 희생된 사람은 약 110만명으로 추산하고 있다[1].
1918년의 인플루엔자 대유행의 바이러스를 이용하여 만들어진 백신이 1957년의 인플루엔자를 예방하지 못한 것은 1957년의 바이러스는 기존에 알려진 인플루엔자 바이러스와는 상당한 차이가 있어서 기존의 백신에 의해서 형성된 항체가 효과적으로 무력화시킬 수 없었기 때문이다.
1970년대에 이르러 바이러스의 아미노산을 분석할 수 있는 기술이 나와서 바이러스를 분석해 본 결과, 이 바이러스는 오리에서 발견된 인플루엔자 바이러스와 유사성을 보였다. 현재 이 인플루엔자 바이러스는 H2N2 서브타입으로 구분되고 이는 1918년의 대유행을 몰고온 H1N1 서브타입과는 구분되고 있다.
1957년의 H2N2 인플루엔자 바이러스 역시 여기에 대응하는 백신이 만들어졌고, 백신의 개발 덕분으로 1957년에 시작된 인플루엔자 대유행은 전세계적으로 수천만명에서 1억명에 달하는 사망을 낸 1918년의 인플루엔자에 비해서는 상대적으로 적은 피해를 내고 끝나긴 했지만, 인플루엔자 대유행의 재현은 인플루엔자가 백신의 개발만으로 완벽히 근절되기 어려운 질병이라는 것을 일깨워주게 되었다.
이러한 인플루엔자 대유행은 주로 가축 유래의 인플루엔자 바이러스가 인간에게 전파되는 방식으로 그 이후에도 계속 일어났다. 1968년경에는 홍콩에서 H2N2 바이러스의 변종으로 추정되는 H3N2 바이러스에 의한 대유행이 시작되었고, 이 유행은 전세계로 퍼져나가 미국에서만 약 3만3,800명의 희생자를 냈다.
가장 최근에 전세계적인 인플루엔자 대유행은 2009년의 돼지에서 유래된 것으로 보이는 H1N1 바이러스의 변종에 의해서 일어났으며, 전세계적으로 11%에서 21%에 달하는 인구가 감염되어 약 15만명에서 57만명 사이로 추산되는 희생자를 냈다.
기존의 백신에 의해서 예방되지 못하는 신종 인플루엔자 바이러스가 지속적으로 출현하는 이유는 인플루엔자 바이러스의 복제를 담당하는 인플루엔자 바이러스 유래의 RNA 의존성 RNA 중합효소(RdRp)가 교정 기능이 없어서 높은 돌연변이율을 보이기 때문이다.인플루엔자 바이러스는 DNA 기반 병원체에 비해 약 300배 이상 높은 돌연변이율을 보이는 것으로 추정되고 있다[2]. 이렇게 높은 돌연변이율 때문에 인플루엔바 바이러스는 필연적으로 항원으로 숙주세포에 인식되는 부분이 지속적으로 변하는 항원 부동(Antigenic drift)를 겪고 있으며, 가끔은 두 가지 전혀 다른 인플루엔자 바이러스가 혼합되어 유전자가 융합되어 전혀 새로운 타입의 바이러스가 나오는 항원 대변이(Antigenic shift)가 발생하고, 대부분의 전세계적인 인플루엔자 대유행의 원인은 항원 대변이에 의한 신종 바이러스 때문이다.
결국 인플루엔자 바이러스와의 싸움은 근원적으로 끝나지 않은 싸움일 수 밖에 없고 백신만으로 완벽히 퇴치가 불가능하다. 이러한 상황에서는 인플루엔자 바이러스에 이미 감염된 환자들의 증상을 완화시켜줄 수 있는 항 바이러스 제제의 개발이 필요하다는 인식이 제기되기 시작하였다. 그러나 이를 위해서는 먼저 인플루엔자 바이러스가 어떤 과정을 통하여 증식하는지에 대한 이해가 선행되어야만 했다.
뉴라미니데이즈
인플루엔자 바이러스의 외피에는 헤마글루티닌(Hemaglutinin)과 뉴라미니데이즈(Neuraminidase)라는 두 개의 당단백질이 있다. 헤마글루티닌은 침투하는 세포막에 존재하는 글리칸의 시알산(Sialic acid)에 결합하여 바이러스를 세포 내로 융합시키는데 필수적인 역할을 한다. 이렇게 바이러스가 세포 내에 침투하는데는 시알산은 필수적이지만, 시알산이 계속 세포막에 존재하면 복제된 바이러스가 숙주세포를 떠날 때 이를 방해한다. 따라서 시알산을 제거하여 세포 밖으로 이탈한 바이러스가 다시 융합되지 않도록 하는 것이 필요하며, 이 작업은 인플루엔자 바이러스 외피에 존재하는 뉴라미니데이즈에 의해서 이루어진다.
따라서 뉴라미니데이즈가 기능을 제대로 하지 못하면 바이러스의 증식이 불가능하며, 뉴라미니데이즈를 항바이러스 약물의 타깃으로 쓸 수 있을 것이라는 가능성이 제시되기 시작하였다. 이러한 시도는 1960년대부터 시작되었다. 가령 1966년 영국의 영국의 에딘버러 제약산업(Edinburg Pharmaceutical Industry)의 연구자들은 시험관 내에서 인플루엔자 바이러스의 뉴라미니데이즈의 효소 활성을 저해하는 옥사믹산(Oxamic acid) 계열의 화합물을 발굴하였다[3]. 그러나 이 화합물은 수정란에서 배양되는 바이러스에 처리하였을때 빠르게 분해되어 약물로서의 가치는 전혀 없었다.
1974년 뉴라미니데이즈가 만드는 시알릭산 유사체 기반의 새로운 뉴라미니데이즈 저해물질이 발굴되었다[4]. 그러나 이 물질은 1M 정도의 비교적 낮은 저해능력을 가진 물질이었고, 세균과 동물 유래의 뉴라미니데이즈 등도 같이 저해하는 비특이적인 화합물이라서 약물로 개발될 가치는 크지 않았다.
바이러스 뉴라미니데이즈를 특이적으로 저해하는 물질을 개발할 수 있게 된 결정적인 계기는 1983년 오스트레일리아 생체분자연구소(Biomolecular Research Institute)의 피터 콜만(Peter M Colman)이 인플루엔자 바이러스 H2N2 서브타입 유래의 뉴라미니데이즈의 단백질 결정 구조를 규명한 이후부터였다[5]. 규명된 뉴라미니데이즈의 구조는 4개의 서브유니트가 마치 4개의 꽃잎으로 구성된 것처럼 복합체를 형성하고 있었고, 각각의 서브유니트의 중앙에는 기질이 결합할 수 있을 만한 결합 포켓이 존재하고 있었다. 1991년 규명된 시알산과 뉴라미니데이즈의 복합체 구조를 통하여 시알산이 결합하는 뉴라미니데이즈의 아미노산이 파악되었으며[6], 이들은 인플루엔자 A와 B 바이러스 변종에서 거의 변화가 없는 보존된 아미노산들이었다. 이러한 뉴라미니데이즈의 구조 정보에 기반하여 뉴라미니데이즈를 저해할 수 있는 약물을 설계하고자 하는 노력이 진행되었다.
최초의 구조 기반 뉴라미니데이즈 저해제, 자나미비르
오스트레일리아의 모나시 대학(Monash University), 최초로 뉴라미니데이즈 구조를 규명한 생체분자연구소 및 오스트레일리아의 바이오텍 기업인 바이오타(Biota)는 뉴라미니데이즈 구조 정보를 이용하여 이를 저해하는 시알산 유사체 기반의 뉴라미니데이즈 저해제 개발을 시도하였다. 이들은 시알산과 뉴라미니데이즈와의 결합 구조를 이용하여 결합된 리간드와 단백질간의 결합력을 분석하는 컴퓨터 프로그램을 사용하여 시알산의 4번째 히드록시기를 아미노기로 바꾸는 것이 가장 안정한 결합을 이룬다는 것을 알아내었고, 이를 기반으로 4번째 히드록시기를 아미노기(-NH3) 혹은 구아니디노기(-HNC(NH2)2)로 바꾸니 nM 대의 Ki 를 가지는 강력한 저해제를 만들 수 있었다[7]. 화합물과 뉴라미니데이즈의 복합체 구조 분석 결과 화합물에 달린 아미노기 혹은 구아니디노기는 뉴라미니데이즈의 활성 자리에 있는 119번 글루탐산 잔기와 이온 결합을 하게 되었고, 이것이 이 화합물의 강력한 저해 능력의 원인이라는 것이 밝혀졌다.
이 화합물은 세포 배양 시스템에서 인플루엔자 바이러스 A, B 의 증식을 모두 억제하는 것이 발견되었으며, 동물 모델에서도 항바이러스 활성이 입증되었고 이 화합물은 자나미비르(O)라는 이름을 가지게 되었다. 1990년, 글락소(Glaxo, 현재 글락소스미스클라인 GSK)는 이 화합물의 권리를 취득하였다. 1999년 FDA는 자나미비르의 판매를 승인하였고, 자나미비르는 리렌자(Relenza)라는 이름으로 판매되기 시작하였다. 리렌자의 경우 경구 생체이용율(oral bioavailabity)가 떨어지고, 빠르게 배출되는 이유로 경구투여제로 만들지 못하였고, 대신 분말 형태의 흡입제 형태의 제형의 약물이 되었다.
리렌자는 뉴라미니데이즈를 저해하는 약물로써 최초로 등장한 퍼스트 인 클래스(First-in class)의 저해제였으나 이것이 시장에서 독점적인 위치를 차지할 수 있는 시간은 몇 개월에 지나지 않았고, 곧 이어 출시된 다른 약물의 도전을 받게 된다.
오셀타미비르(타미플루)의 등장
길리아드(Gilead Science) 는 항바이러스제제 개발에 특화된 바이오텍으로써 뉴라미니데이즈를 저해하는 화합물의 개발에 착수하게 된다. 이들은 자나미비르가 경구 투여가 불가능하고 흡입에 의해서만 투여 가능하다는 것에 힌트를 얻어 시알산 유사체가 아닌 새로운 스캐폴드를 이용한 약물의 개발을 시도하였다. 이들은 뉴라미니데이즈의 반응 중간체(Transition state) 와 유사한 입체적 구조를 가지는 사이클로헥센(Cyclohexene) 을 스캐폴드로 사용하여 뉴라미니데이즈와의 복합체 구조를 참조하여 화합물을 개발하였다. 화합물의 최적화 과정에서 사이클로헥센의 3번쨰 탄소에 결합하는 잔기를 좀 더 소수성인 것으로 바꿀수록 뉴라미니데이즈의 저해 활성이 높아진다는 것을 발견하였다. 이를 통하여 인플루엔자 A와 B 유래의 뉴라미니데이즈를 1-3nM 수준으로 저해하는 강력한 후보 물질인 GS 4071 이 도출되었다[8].
그러나 GS 4071의 경우 래트에서의 경구 이용률이 5%에 지나지 않았다. 원래의 개발 목적인 생체이용율을 높여서 경구 투여가 가능하는 목적에 부응하지 않는 결과였다. 이를 개선하기 위하여 GS4071의 카르복시기에 에틸에스테르(Ethyl ester)를 결합시켜 생체 내에서 활성이 있는 형태로 변환될 수 있는 프로드럭(Prodrug) 형태의 화합물이 만들어졌고 이는 GS4104라는 이름이 붙여졌다[9]. 이 화합물은 원래의 화합물인 GS4071에 비해서 경구 생체이용율이 5배 이상 증가하여 원래의 개발 목적인 경구 투여가 가능하게 되었다.
GS4014는 오셀타미비르(Osteltamivir)라는 정식 성분명을 얻었으며, 1999년 FDA로부터 판매 허가를 얻어 ‘타미플루’(Tamiflu)라는 상품명으로 팔리기 시작했다. 오셀타미비르는 흡입제 형태로만 투여가 가능한 자나미비르에 반해 경구 투여가 가능한 최초의 뉴라미니데이즈 저해제였으며, 타미플루는 리렌자에 비해서 높은 시장 점유율을 가지게 되었다.
그러나 타미플루의 경우 뉴라미니데이즈에서 발생하는 돌연변이에 의해서 저항성을 가지는 바이러스가 출현한다는 것이 보고되기 시작하였다. 이러한 돌연변이중의 하나인 H1N1 인플루엔자 바이러스 뉴라미니데이즈의 275번째 히스티딘이 타이로신으로 바뀐 돌연변이는 2017년의 조사 결과 약 1% 의 바이러스에서 발견되었다[10]. 반면 리렌자의 경우 현재까지 여기에 저항성이 있는 바이러스가 출현된 보고가 없는 관계로 타미플루에 내성을 가지는 환자들에게는 처방이 권장되고 있다.
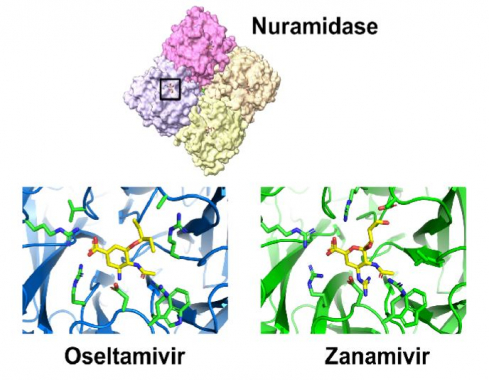
▲그림 : (상) 인플루엔자 바이러스의 뉴라미니데이즈(Neuraminidase)의 구조. 뉴라미니데이즈는 동일한 서브유니트가 4개 결합한 구조로 되어 있으며, 각각의 서브유니트 중앙마다 활성자리 (Active Site)가 존재한다. (하) 오셀타미비르 (Oseltamivir : 타미플루) 와 자나미비르 (Zanamivir : 릴렌자) 가 뉴라미니데이즈의 활성자리에 결합되어 있는 모습. 각각의 화합물은 노란색으로 표시되어 있으며, 이와 상호작용하는 아미노산 잔기는 초록색으로 표시되어 있다.
다음 연재에서는 C형 간염 바이러스 치료제의 개발 과정을 알아봄으로써 10회에 걸친 인간의 바이러스와의 전쟁의 역사를 마무리하고자 한다.
참고문헌
Viboud, C., Simonsen, L., Fuentes, R., Flores, J., Miller, M. A., & Chowell, G. (2016). Global mortality impact of the 1957–1959 influenza pandemic. The Journal of infectious diseases, 213(5), 738-745.
Drake, J. W. (1993). Rates of spontaneous mutation among RNA viruses. Proceedings of the National Academy of Sciences, 90(9), 4171-4175.
Edmond, J. D., Johnston, R. G., Kidd, D., Rylance, H. J., & Sommerville, R. G. (1966). The inhibition of neuraminidase and antiviral action. British journal of pharmacology and chemotherapy, 27(2), 415.
Meindl, P., Bodo, G., Palese, P., Schulman, J., & Tuppy, H. (1974). Inhibition of neuraminidase activity by derivatives of 2-deoxy-2, 3-dehydro-N-acetylneuraminic acid. Virology, 58(2), 457-463.
Varghese, J. N., Laver, W. G., & Colman, P. M. (1983). Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature, 303(5912), 35–40. doi:10.1038/303035a0
Burmeister, W. P., Ruigrok, R. W., & Cusack, S. (1992). The 2.2 A resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. The EMBO journal, 11(1), 49-56.
von Itzstein, M., Wu, W. Y., Kok, G. B., Pegg, M. S., Dyason, J. C., Jin, B., ... & Colman, P. M. (1993). Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature, 363(6428), 418.
Lew, W., Chen, X., & Kim, C. U. (2000). Discovery and development of GS 4104 (oseltamivir) an orally active influenza neuraminidase inhibitor. Current medicinal chemistry, 7(6), 663-672.
Li, W., Escarpe, P. A., Eisenberg, E. J., Cundy, K. C., Sweet, C., Jakeman, K. J., ... & Kim, C. U. (1998). Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrobial Agents and Chemotherapy, 42(3), 647-653.
https://www.cdc.gov/flu/treatment/antiviralresistance.htm





![[인사]일동제약, 신임 R&D 본부장에 박재홍 사장 선임](https://img.etoday.co.kr/crop/268/200/2316073.jpg)

