기사본문
[남궁석의 신약연구史]질병관련 단백질 구조의 규명
입력 2022-05-13 10:23 수정 2022-05-15 10:59
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
분자생물학과 구조생물학의 기술이 확립된 이후 이제 많은 연구자들은 인간의 질병에 관련된 많은 단백질의 구조에 관심을 가지게 됐다. 결국 약물은 특정한 단백질에 작용하여 효과를 내고, 질병에 직접적으로 연관된 단백질의 구조를 알게되면 질병의 치료법을 찾을 수 있다는 기대로 질병 단백질의 구조에 대한 연구가 시작됐다. 물론 이러한 기대는 바로 이루어진 경우도 있었지만, 대개는 단백질 구조가 밝혀진 이후에도 해당하는 단백질을 표적화하는 치료제가 나오는데는 오랜시간이 걸리는 경우가 많았다. 그러나 질병 관련 단백질의 구조는 질병의 기본적인 메커니즘에 대한 우리의 이해를 깊게 한 것은 분명하다. 이번 연재에서는 구조생물학이 질병에 관련된 단백질의 구조를 규명하는 과정을 알아보도록 한다.
암 단백질 라스의 구조
1980년대 분자생물학의 발전 이후 많은 연구자들이 관심을 가진 것은 암이 어떻게 일어나는지에 대한 분자적 근원이었다. 이후 유전자에 돌연변이가 생겨서 기능이 조절되지 않고 활성화가 계속되면 암 성장을 촉진하는 ‘암 유전자(Oncogene)', 혹은 정상세포에서는 유전정보의 손상을 억제하고 있지만, 만약 돌연변이가 생겨서 기능을 잃으면 유전정보의 손상이 가속화되어 암 유발 확률을 높이는 ‘암 억제 유전자(Tumor Suppressor)'들이 발견되기 시작했다.
‘암 유전자'로 가장 처음 알려진 유전자 중의 하나가 바로 ‘K-ras’다. 오늘날 고형암의 20% 정도에서 발견되는 돌연변이 K-ras 유전자는 원래 쥐에서 암을 일으키는 바이러스에 의해서 전파되는 유전자로 처음 발견됐다. 이와는 별도로 돌연변이에 의한 DNA 손상이 암을 유발한다고 믿었던 연구자들은 방광암에서 정상세포를 암으로 바꾸는 DNA 조각을 발견했고, 이것이 이전에 바이러스에 발견된 K-ras와 동일한 유전자라는 것이 발견됐다. 그리고 암세포에서 발견된 K-ras 유전자는 정상세포에도 존재하는 유전자에 단 하나의 염기변화가 존재하는 돌연변이 유전자였다.[1]
그렇다면 이러한 돌연변이는 어떻게 단백질에 영향을 미쳐 암을 유도할까? 해당 단백질의 3차원 구조를 알게 된다면 어떻게 돌연변이가 단백질의 기능에 영향을 미쳐, 암을 유도하는지에 대해서 좀 더 잘 이해할 수 있게 될 것이다.
1988년 캘리포니아 대학의 구조생물학자 김성호(Sung-Hou Kim, 1937-)는 ras 단백질의 구조를 규명했다.[2] 그러나 의외로 암세포에서 발견되어 암을 유발하는 돌연변이를 가지고 있는 돌연변이 ras 단백질의 구조는 돌연변이가 없는 단백질과 다르지 않았다. 그렇다면 이 차이를 어떻게 설명할 수 있을까? 이러한 의문은 1990년 GTP가 결합된 ras 단백질과 GDP가 결합된 ras 단백질의 구조를 풀면서 풀렸다.[3] ras는 GTP 혹은 GDP를 결합하는데, 어떤 기질을 결합하느냐에 따라서 그 구조가 크게 달라진다. GTP가 결합된 ras는 다른 단백질과 결합하여 세포 증식을 계속하라는 신호를 보내지만, GDP가 결합한 ras는 구조가 달라지면서 세포증식 계속의 신호를 더 이상 보낼 수 없다. 즉 ras라는 단백질은 세포내에서 GTP와 GDP가 결합한 상태를 서로 오가면서 세포가 증식하는 것을 조절하는 스위치의 역할을 한다.
그러나 ras 유전자에 암을 유발하는 돌연변이가 있으면 상황이 달라진다. 가령 암 세포에서 발견된 ras 유전자는 12번째 아미노산인 글리신이 시스테인(G12C), 혹은 아스파르트산(G12D) 등으로 변형되어 있는 경우가 많다. 이렇게 돌연변이가 있는 ras 단백질은 붙어 있는 GTP가 GDP로 교환되지 않고 그대로 GTP가 붙어 있게 된다. 즉, 돌연변이가 있는 ras 단백질은 세포증식을 유도하는 GTP가 결합된 구조로 항상 남아 있기 때문에 스위치가 항상 켜져있는 상태로 있고, 세포증식이 필요하지 않은 상태에서도 세포증식을 유도하게 된다. 암이 발생되는 이유가 세포증식이 조절되지 않고 무절제하게 세포가 증식하는 것이라는 것을 생각한다면, ras 단백질의 구조 규명은 왜 ras 유전자의 돌연변이가 암을 유발하는지를 설명하는데 핵심적인 기여를 한 셈이다.
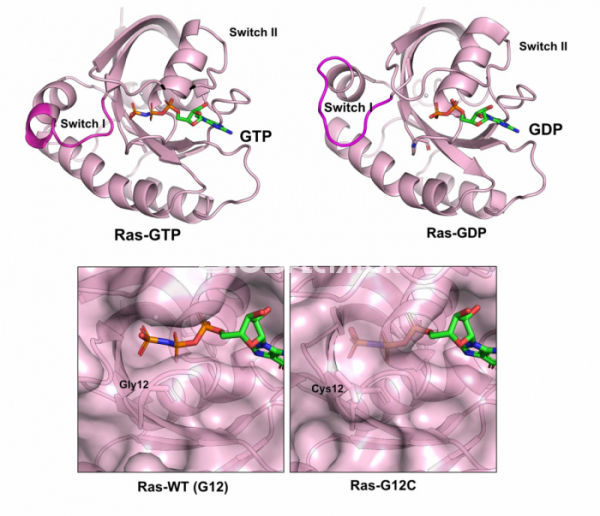
▲그림 1. Ras-GTP와 Ras-GDP의 구조. Ras는 GTP혹은 GDP에 결합하느냐에 따라서 구조가 달라지는 단백질이며, GTP에 의해서 구조가 변하는 부분은 스위치 I(Switch I) 및 스위치 II(Switch II)라고 부른다. GTP가 붙어있는 Ras는 세포의 성장 신호를 전달하는 매개체가 된다. 정상 세포에서 Ras는 세포의 성장 신호에 따라서 GTP와 GDP를 오가며 서로 다른 상태에 있게 된다. 그러나 암세포에서는 Ras유전자에 돌연변이가 빈번하게 발생하며, 12번째 글리신이 시스테인으로 바뀐 (G12C) 등의 돌연변이가 생기면 Ras는 GTP 상태에 그대로 남아 있게 되어 세포의 성장 신호를 항상 보내게 되고 이는 암 세포의 지속적인 성장을 유도한다. 아생형 Ras (Ras-WT)에 붙어있는 인산기는 외부로 노출되어 쉽게 분해되지만, Ras-G12C의 GTP의 인산기는 Ras 단백질 내부에서 보호되어 쉽게 가수분해되지 않음을 구조를 통해 알 수 있다.
물론 이러한 발견이 당장 항암 치료제의 개발로 이루어지지는 않았다. 실제로 돌연변이 ras 단백질에 결합하여 암 세포의 증식을 막는 물질은 ras 단백질의 구조가 밝혀진 후 약 30년이 넘은 뒤에야 개발됐다. 그러나 이러한 개발에는 ras 단백질의 구조와 이의 조절기전의 규명이 지적 기반이됐다.
단백질 인산화 효소의 구조
1970-1980년대 암 유전자를 연구하던 연구자들은 암을 유발하는 유전자의 상당수가 돌연변이가 생긴 단백질 인산화효소(Protein Kinase)임을 깨닫기 시작했다. 1976년 해롤드 바무스(Harold E Varmus)와 마이클 비숍(Micheal J Bishop)은 이전부터 조류세포에서 암을 유발하는 바이러스로 알려져 있던 라우스 사코마 바이러스(Rous Sarcoma Virus)에서 암을 유발하는 유전자는 정상적인 조류 세포에도 있는 유전자지만, 다만 C말단의 몇개의 아미노산이 결여되어 있는 돌연변이 유전자라는 것을 알게 됐다. v-Src라고 불리는 이 유전자는 단백질의 타이로신 잔기에 인산기를 달아주는 타이로신 단백질 인산화효소(Tyrosine Protein Kinase)였다. 한편 정상 세포에 들어있는 c-Src라는 단백질에 추가된 부분은 단백질 인산화효소 활성의 ‘브레이크' 역할을 해 주게 된다. 즉, c-Src 단백질 역시 세포가 증식하느냐 마느냐를 결정하는 신호를 전달해 주는 역할을 하게 되는데, 암을 유발하는 v-Src 단백질은 단백질 인산화효소 활성의 브레이크가 결여되어 있어서, 항상 ‘스위치가 켜져 있는' 상태가 되고 이렇게 계속 세포 증식의 신호가 켜져 있는 돌연변이 단백질이 암의 원인이 되는 것이다.[4]
비슷한 시기에 암의 원인으로 규명된 유전자 중의 하나는 만성 골수성 백혈병(Chronic Myeloide Leukimedia, CML)를 유발하는 유전자다. 1960년대에 만성 골수성 백혈병 환자의 암세포에는 비정상적으로 작은 염색체가 있다는 것이 밝혀졌고, 이후에 이것은 9번 염색체와 22번 염색체가 일정 부위에서 절단되어 서로 이동되기 때문에 형성되는 것으로 밝혀졌다. 1980년대에 이르러 이러한 염색체 치환이 9번 염색체에 있던 ‘ABL’이라는 유전자와 22번 염색체에 있는 ‘BCR’이라는 유전자의 융합을 만들고, 두 개의 단백질이 합쳐진 융합 단백질을 만든다는 것이 알려졌다. 그렇다면 이렇게 융합 단백질이 만들어지는 것과 암 발생은 어떤 관계가 있을까? ABL 역시 단백질 인산화효소 유전자였고, BCR과 ABL 유전자가 융합되면, BCR-ABL이라는 정상적으로 존재하지 않는 융합 단백질이 만들어지고, 이 경우 단백질 인산화효소인 ABL의 활성이 조절되지 않고, 항상 활성이 유지된 상태로 존재하게 된다. ABL이 항상 활성을 유지한 상태로 있게 되면 세포성장 신호가 계속 켜져있게 되고, 백혈병 암세포는 무절제하게 증식하게 된다. Ras의 경우와 마찬가지로 많은 단백질 인산화효소 역시 활성을 조절하는 스위치가 망가져서 항상 활성이 켜져있게 되면 암의 원인이 되는 것이다.
이렇게 여러가지 단백질 인산화 효소의 문제로 암이 발생한다는 것이 알려진 이후, 단백질 인산화효소를 화합물로 저해하면 암을 치료할 수 있지 않을까 하는 아이디어가 제시됐다. 그러나 문제는 우리 몸에 존재하는 단백질 인산화효소는 모두 500개에 달하고, 이들은 모두 유사한 서열을 가진 단백질 인산화효소 도메인을 가지고 있다는 것이었다. 단백질 인산화를 담당하는 도메인이 모두 유사하다면, 단백질 인산화효소의 구조는 대개 비슷할 것이다. 그리고 약 500개에 달하는 단백질 인산화효소는 세포증식뿐만 아니라 우리 몸에서 다양한 조절기전에 관련되어 있다. 만약 단백질 인산화효소를 저해하는 화합물이 있다고 하더라도 목적하는 단백질 인산화효소 이외의 다양한 단백질 인산화효소를 저해한다면 독성이 야기될 것이고, 그렇다면 약물로 과연 쓸모있을 것인가?
이러한 의문이 제기되는 와중에 1991년 최초의 단백질 인산화효소의 구조인 단백질 인산화효소 A(Protein Kinase A)의 구조가 규명됐다.[5] 규명된 단백질 인산화효소 A의 구조는 크게 N말단에 존재하는 베타 쉬트로 주로 이루어진 영역(N-lobe)와 C말단에 위치한 주로 알파 나선으로 이루어진 영역(C-lobe)로 구성된 2개의 도메인이 결합되어 있는 형태로 되어 있었으며, 단백질의 인산화가 이루어지는 장소는 가운데 부분이고 여기에 인산기를 전달하는 ATP가 결합되어 있었다. 단백질 인산화효소 A는 타이로신 대신 세린 혹은 쓰레오닌에 인산화를 하는 세린/쓰레오닌 단백질 인산화효소이며, 생체 내에서 에너지 대사에 관련된 단백질의 기능을 조절하는 역할을 한다. 1994년 최초의 타이로신 단백질 인산화효소의 구조인 인슐린 수용체 단백질 인산화효소의 구조가 밝혀졌으며, 이의 구조 역시 세린/쓰레오닌 단백질 인산화효소와 크게 다르지 않음을 알 수 있었다.[6]
그렇다면 타이로신 인산화효소의 활성은 어떻게 조절될까? 1996년 인간 림프구 인산화효소 (Human Lymphocyte kinase, LCK)라는 타이로신 인산화효소의 활성 상태에서의 구조가 풀렸다.[7] 인간 림프구 인산화효소는 c-Src와 유사한 조절기전을 가지고 있는 타이로신 인산화효소로써, C말단에 존재하는 타이로신 잔기에 인산화가 되어 있으면 활성이 없어지지만, C말단의 타이로신 잔기의 인산기가 사라지고, 인산화효소 가운데에 존재하는 394번 타이로신에 인산화가 되면 활성화가 되는 것으로 알려져 있었다. 이 구조를 기존에 풀린 인슐린 수용체 단백질 안산화효소와 비교해 보니 활성화된 인산화효소는 인산화될 단백질이 붙는 활성자리에 가까운 부분이 열려 있는 상태인 반면, 기존의 인슐린 수용체 단백질 인산화효소는 활성자리 부분의 영역이 닫혀 있는 형태로 되어 있음을 알 수 있었다. 즉 타이로신 인산화효소의 활성은 ‘활성화 루프(Activation Loop)'라고 불리는 영역이 열려서 단백질이 활성자리에 결합되느냐 아니냐로 우선 결정되는 것이다.
그리고 c-Src과 같이 단백질의 C말단에 조절 영역이 존재하는 단백질 인산화효소는 어떻게 조절되는가? 이러한 의문은 1999년에 c-Src의 구조가 풀리면서 해결됐다.[8] C말단의 타이로신이 존재하는 조절 영역의 타이로신은 인산화된 상태에서 단백질의 N말단에 있는 SH2 도메인에 결합하고, 이렇게 단백질 내에서 일어나는 상호작용에 의해서 타이로신 인산화효소의 구조는 비활성화된 상태로 유지된다. c-Src의 조절 영역이 없어진 바이러스의 돌연변이 v-Src 단백질은 활성화된 상태로 유지되기 때문에 항상 세포의 증식 신호를 보내고, 이는 암세포의 증식으로 이어지게 되는 것이다.
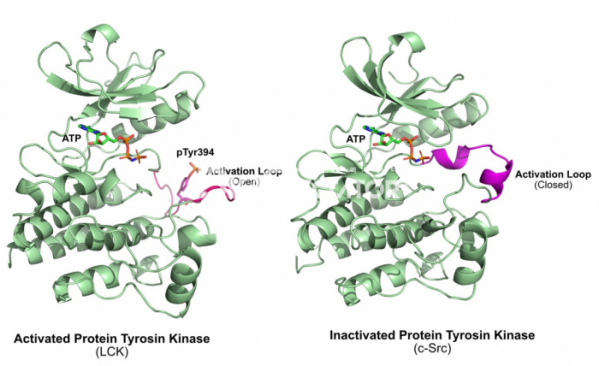
▲그림 2. 활성화된 단백질 타이로신 인산화효소(Lck)와 비활성화된 단백질 인산화 효소(c-Src)의 비교. 394번 타이로신이 인산화된 LCK의 활성화 루프(Activation Loop)는 바깥쪽으로 돌출되어 있어 인산화될 단백질의 결합을 용이하게 한다. 그러나 비활성화된 상태에서 활성화 루프는 알파 나선을 형성하여 기질의 결합을 막게 된다. 상당수의 단백질 인산화효소는 이렇게 인산화에 의해서 활성화 또는 비활성화되어 그 기능이 엄격히 조절된다.
암 억제 단백질의 구조
암 유전자 이외에 중요한 암과 관련된 유전자에는 DNA의 손상을 억제하여 암 발생을 억제해 주는 ‘암 억제 유전자'가 있다는 것은 이미 이야기했다. 그렇다면 이 암 억제 유전자에서 생성되는 암 억제 단백질은 세포 내에서 실제로 어떤 기능을 수행하고 있을까?
암 억제 단백질의 대표적인 단백질이자 암에서 가장 빈번하게 돌연변이가 일어나는 단백질인 p53은 1979년 일련의 연구자들에 의해 발견됐다. 이후의 연구를 통하여 p53은 세포의 DNA 손상을 막아서 세포가 암세포로 변하는 것을 막아주는데 핵심적인 역할을 한다는 것이 밝혀졌다. 만약 세포가 유해성 물질이나 자외선 등에 노출되어 DNA 가 손상되면, 이를 그대로 두면 세포의 DNA에 돌연변이가 누적되고, 유전 정보의 손상이 계속되다 보면 세포의 성장을 조절하는 암 유전자 등에 돌연변이가 생겨 암이 유발된다. 만약 DNA 손상이 수선 가능한 수준이라면 p53은 DNA의 수선에 관여하는 유전자들의 발현을 촉진시켜 DNA 수선을 유도한다. 그러나 DNA 손상이 수선 가능한 수준을 넘어선다면, 더 이상의 세포 증식을 억제하고 세포 사멸을 유도하여 개체의 유전 정보 손상을 막는다.
p53이 다른 유전자의 발현을 촉진하기 위해서는 일단 DNA에 결합하여야 하고, p53 단백질 내에서 DNA에 결합하는 부분은 단백질의 가운데 위치하는 약 200 아미노산으로 이루어진 DNA 결합 도메인 부분이다. 1994년 미국 뉴욕의 슬론케터링 암센터의 니콜라 파블레비치(Nicolas Pavletich)연구실에서는 p53의 DNA 결합 부위와의 구조를 규명했다.[9] 단백질-DNA의 복합체 구조와 암 환자에서 발견된 p53 유전자 내의 돌연변이를 연결지어 관찰하니 매우 흥미로운 관계가 도출되었다. 즉, 암 환자의 P53에서 가장 빈번하게 돌연변이가 발생하는 부분은 바로 DNA 결합 도메인이며, DNA 결합 도메인의 아미노산 중에서도 직접 DNA의 인산기와 상호작용하는 아미노산들인 175,248,249, 273 번째 아르기닌에서 돌연변이가 가장 많이 발생하였다. 결국 P53의 DNA 결합 부위에 돌연변이가 발생하여 p53 단백질이 더이상 DNA에 결합하지 못하기 때문에 암 억제 유전자로서의 기능이 망가지는 셈이다.
p53은 DNA 손상이 많이 될 때 DNA 수선, 세포증식 억제, 세포사멸을 유도하는 단백질이므로 DNA 손상이 없는 정상상태에서는 작동해서는 안된다. 그렇다면 P53의 기능은 어떻게 조절될까?
p53을 연구하던 과학자들은 정상 상태에서는 P53은 MDM2(MDMX)이라는 단백질과 결합하여 있고, 이로 인해 P53의 전사 활성화 기능이 저해된다는 것을 알게 되었다. 1996년 파블레비치 연구실에서는 MDM2 단백질과 p53의 전사 활성화 도메인의 복합체 구조를 밝혔다.[10] 이를 통해 MDM2 단백질은 p53이 DNA에 결합할 때 유전자의 전사를 활성화하는데 필요한 부분에 경쟁적으로 결합하는 저해 단백질로 작용한다는 것을 알 수 있었다.
이와 더불어 MDM2 은 p53에 유비퀴틴을 결합시켜 단백질을 분해시키는 유비퀴틴 라이게이즈로써의 역할도 하고 있다. 즉 p53에 결합하여 이의 전사 활성화 기능을 억제시켜며, 동시에 p53 을 분해시키는 두 가지 기작을 통하여 p53이 정상 상태에서는 기능을 못하도록 강력히 억제하고 있는 것이다. 그러나 만약 MDM2가 지나치게 많이 존재하여 p53이 기능해야 할 상황에서도 p53의 기능이 억제되어 버리면 어떻게 될까? 많은 암 환자에게서 MDM2 유전자에 변형이 생겨서 MDM2 유전자가 너무 과다하게 만들어져 버리고, 따라서 DNA 손상이 일어나 p53이 필요할 때 기능을 못하고, 따라서 암 발생이 촉진되는 경우가 발생한다. 이렇게 MDM2가 비정상적으로 많이 존재하는 경우 MDM2 와 p53이 결합하는 것을 막을 수 있다면 p53의 기능을 다시 회복할 수 있을지도 모른다. 이러한 가능성은 1990년대 말 MDM2 와 p53간의 복합체 구조가 규명되었을때 제시되었으나, 이 가능성이 실제로 약물로 등장하기까지는 역시 많은 시간이 흘러야만 했다.
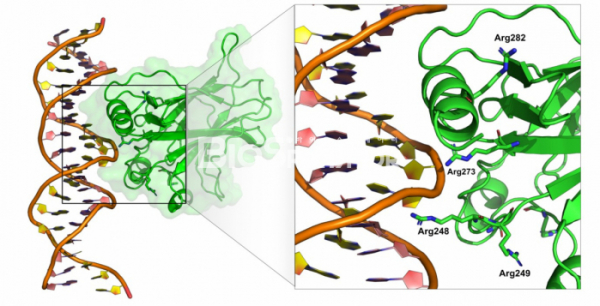
▲그림 3. p53과 DNA의 결합 구조. 암 억제 유전자 p53은 DNA에 결합하여 DNA 손상 수리에 관련된 유전자의 발현을 유도하며, 암 환자에게서 가장 빈번히 돌연변이가 나타나는 유전자이다. P53과 DNA의 결합 구조를 규명한 이후, p53에서 가장 빈번하게 돌연변이가 나타나는 부분이 바로 p53과 단백질과 직접 결합하는 273, 282, 248번째 아르기닌 등의 아미노산이다.
바이러스 유래의 단백질의 구조
1980년대 암 관련 유전자의 발굴이 한창 진행될 때쯤 이와 함께 크게 이슈가 된 것은 인간면역결핍바이러스(Human Immunodifiency Virus, HIV)의 감염에 의해서 유발되는 후천성면역결핍증, 즉 에이즈(AIDS)의 창궐이었다. 우리 몸의 면역 콘트롤타워인 헬퍼 T 세포에 감염되어, 헬퍼 T 세포의 숫자를 점차 줄임으로써, 거의 모든 적응성 면역력을 서서히 감소시키는 에이즈는 한때 ‘20세기의 흑사병'으로 여겨지면서 인류종말을 가져올 위협적인 질병으로 여겨졌다. 그러나 1990년대 중반, ‘고활성 항레트로바이러스 요법(HAART, highly active antiretroviral therapy)'이라고 불리는 새로운 항 바이러스 치료법이 등장하면서 에이즈는 서서히 인류의 종말을 가져울 수 있는 질병에서, 지속적으로 약을 복용할 경우 오랫동안 증상없이 관리 가능한, 고혈압이나 당뇨와 같은 만성질병의 위치로 변하게 되었다. 물론 아직도 에이즈는 전세계적으로 아프리카와 같이 보건 인프라가 부족한 국가에서 1년에 수십만명의 사망자를 내는 치명적인 질병인 것은 사실이지만, 적어도 치료법이 없는 불치의 질병이던 상황과는 현저히 달라지게 되었다.[11]
그렇다면 어떻게 바이러스와 이로 인한 질병이 확인된 지 10여년만에 신속하게 이를 치료할 수 있는 효과있는 약물이 나올 수 있었을까? 여기에는 여러가지 요인이 있었겠지만, 1980년대 이후 급속하게 발전된 분자생물학에 의해 HIV의 유전자들과 그 단백질들의 기능이 속속들이 조사되고, 바이러스의 생활사가 연구됨으로써, 바이러스의 ‘약점'이 드러났고, 이러한 약점을 공략하는 약물이 등장할 수 있었다. 특히 구조생물학의 발전에 의해서 HIV 바이러스의 거의 모든 단백질의 구조가 밝혀졌고, 이렇게 밝혀진 단백질 구조는 단백질을 저해할 수 있는 약물의 개발에 크게 기여했다.
HIV에는 총 15개의 단백질이 존재한다. 이중 바이러스의 본체를 구성하는 구조단백질은 일단 3개의 유전자(gag, pol, env)로부터 3개의 긴 단백질로 일단 번역된 이후, 각각의 긴 단백질은 단백질 분해효소에 의해서 잘려서 다양한 기능을 하는 단백질을 형성한다. 즉 HIV를 구성하는 단백질을 완성하기 위해서는 단백질 분해효소에 의한 절단과정이 필수적이다. 이렇게 HIV의 증식에 필수적인 과정인 관계로 HIV의 단백질 분해효소는 항바이러스 약물의 타깃이 될 수 있을 것이라는 기대를 가졌으며, 단백질 분해효소를 저해하는 약물을 개발하는데 가장 중요한 정보는 역시 단백질 분해효소의 단백질 구조였다.
1989년 미국 제약사 머크(MSD)의 연구진들은 HIV의 단백질 분해효소의 구조를 규명하였으며, 이것은 HIV 유래의 단백질로는 가장 처음 알려진 구조였다.[12] HIV의 단백질 분해효소는 99개의 아미노산으로 구성되어 있고, 활성자리에 아스파르트산이 위치한 ‘아스파르트산 단백질 분해효소’인데, 보통 300개 이상의 아미노산으로 구성된 동물의 일반적인 아스파르트산 단백질 분해효소에 비해서 약 1/3 정도의 작은 크기였다.
규명된 HIV의 단백질 분해효소는 2개의 단백질 단위체가 대칭으로 이합체를 형성하고 있는 구조였고 2개의 단위체 사이에는 분해될 가닥이 붙을 수 있는 부위가 홈처럼 파여져 있었다. 아스파르트산 단백질 분해효소는 활성자리에 아스파르트산-쓰레오닌-글리신의 아미노산이 있고, 이 3개의 아미노산이 단백질 분해효소의 활성에 결정적인 역할을 하는 것으로 알려져 있었는데, HIV 단백질분해효소에는 25, 26, 27번째 아미노산에 이 3개의 아미노산이 배치되어 있었다. 대칭으로 이합체를 형성한 후 분해될 단백질 사이에 서로 마주보고 있는 두 개의 25번 아스파르트산이 산염기 촉매작용을 수행하여 단백질의 펩타이드 결합을 분해하게 된다. 이러한 HIV 단백질 분해효소의 구조는 곧 여기에 결합하여 활성을 저해하는 약물의 개발을 촉진시키게 된다.
이렇게 질병에 관련된 단백질의 구조가 속속 등장하였고, 이러한 질병 관련 단백질의 구조는 ‘구조 기반 신약개발(Structure-based drug discovery, SBDD)'이라는 신약개발의 새로운 패러다임의 기반이 된다. 다음 연재에서는 질병 관련 단백질 구조가 어떻게 신약 개발의 패러다임을 바꾸게 되는지의 과정을 살펴보고자 한다.
참고문헌
1. Parada, L. F., Tabin, C. J., Shih, C., & Weinberg, R. A. (1982). Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature, 297(5866), 474-478
2. Tong, L., Milburn, M. V., De Vos, A. M., & Kim, S. H. (1989). Structure of ras proteins. Science, 245(4915), 244-244.
3. Milburn, M. V., Tong, L., DeVos, A. M., Brünger, A., Yamaizumi, Z., Nishimura, S., & Kim, S. H. (1990). Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science, 247(4945), 939-945.
4. 남궁석, (2019)암 정복 연대기: 암과 싸운 과학자들. 바이오스펙테이터:서울
5. Knighton, D. R., Zheng, J., Ten Eyck, L. F., Ashford, V. A., Xuong, N. H., Taylor, S. S., & Sowadski, J. M. (1991). Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science, 253(5018), 407-414.
6. Hubbard, S. R., Wei, L., & Hendrickson, W. A. (1994). Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature, 372(6508), 746-754.
7. Yamaguchi, H., & Hendrickson, W. A. (1996). Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature, 384(6608), 484-489.
8. Xu, W., Harrison, S. C., & Eck, M. J. (1997). Three-dimensional structure of the tyrosine kinase c-Src. Nature, 385(6617), 595-602; Xu, W., Doshi, A., Lei, M., Eck, M. J., & Harrison, S. C. (1999). Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Molecular cell, 3(5), 629-638.
9. Cho, Y., Gorina, S., Jeffrey, P. D., & Pavletich, N. P. (1994). Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science, 265(5170), 346-355.
10. Kussie, P. H., Gorina, S., Marechal, V., Elenbaas, B., Moreau, J., Levine, A. J., & Pavletich, N. P. (1996). Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science, 274(5289), 948-953.
11. 남궁석, (2021) 바이러스, 사회를 감염하다, 바이오스펙테이터:서울 p183-207
12. Navia, Manuel A.; Fitzgerald, Paula M. D.; McKeever, Brian M.; Leu, Chih-Tai; Heimbach, Jill C.; Herber, Wayne K.; Sigal, Irving S.; Darke, Paul L.; Springer, James P. (1989). Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. , 337(6208), 615–620. doi:10.1038/337615a0




![[인사]일동홀딩스, 새 대표이사에 최규한 사장 선임](https://img.etoday.co.kr/crop/268/200/2313330.jpg)


