기사본문
[남궁석의 신약연구史]단백질구조, 히트발굴 등서 활용법
입력 2022-07-11 14:10 수정 2022-07-14 14:17
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
지난 회의 연재에서 단백질 구조 정보를 응용하여 개발된 신약의 예를 소개했다. 이들 중 상당수는 '시험관 내 활성 검정(In vitro Screening)' 내지는 '세포 수준의 활성 검정(Cell Level Assay)' 등의 실험방법에 의해서 일단 후보물질이 발굴된 다음, 표적 단백질과의 결합구조를 규명하여 약물 최적화에 단백질 구조 정보가 이용된 것이 많다. 초기 히트(Hits) 발굴은 단백질의 구조 정보와 무관하게 얻어진 것들이 상당수였다. 그러나 단백질 구조 규명이 일반화되고, 많은 약물 표적 단백질의 구조가 풀린 다음에는 초기 히트 발굴단계에서부터 약물 표적 단백질의 구조를 적극적으로 활용하여 보다 적은 시간내에 히트를 발굴하려는 시도가 시작되었다. 이렇게 히트가 발굴된 다음 이를 선도물질(Lead)화하고, 그리고 선도물질을 최적화하는데에도 단백질과 화합물의 결합구조는 매우 중요한 역할을 한다. 이번 회에서는 단백질 구조를 초기 히트 발굴부터 적극적으로 이용할 때 사용되는 여러가지 방법론을 중심으로 오늘날 신약개발에서 어떻게 단백질 구조가 중요한 역할을 하고 있는지 알아보도록 한다.
프래그먼트 기반의 신약개발
1990년대 조합 화학(Combinational Chemistry)과 자동화기기의 발전으로 제약회사들은 수백만 종류의 화합물을 그리 어렵지 않게 만들 수 있게 되었고, 이를 이용하여 수백만 종류 이상의 화합물의 활성을 측정하여 약물 후보물질을 검색하는 '하이스루풋 스크리닝(High-throughput Screening, HTS)'이 일반화되게 되었다. 그러나 이렇게 수많은 화합물을 합성하고 활성을 측정할 수 있게 되었어도 신약개발은 여전히 어렵고 비용이 많이 드는 일이었다. 즉, 수백만-수천만의 화합물의 활성을 측정해도 반드시 유의미한 저해 활성을 가지는 화합물을 얻는다는 보장은 없었으며, 어렵게 얻어진 후보 화합물의 경우에도 독성문제나 약물동역학적인 문제 때문에 약물의 개발이 더이상 진척되지 않는 경우가 많았다. 가능한 많은 후보 화합물 중에서 원하는 활성을 가지는 저해 화합물을 찾는 HTS가 주된 약물개발의 패러다임으로 대두된 1990년 이후 의약화학자들은 실제로 인허가를 통과하여 상용화된 약물들이 가지는 공통적인 특성에도 주목하기 시작하였다.
화이자의 의약화학자인 크리스토퍼 리핀스키(Christoper A. Lipinski)는 2001년 발표된 논문에서 경구투여가 가능한 약물은 공통적으로 일정한 특성을 가진다는 것에 주목했다. 이러한 특성은 다음과 같다.
(1)분자량은 500 달톤 이하 (2)logP <5 (P는 옥타놀과 물 사이에서의 분배계수로써 화합물의 지질 친화도를 의미함) (3)수소 결합 주개가 5개 이하 (4)수소 결합 받개가 10개 이하
이러한 특성들은 ‘리핀스키의 5규칙(Lipinski’s rule of 5)'라고 통칭되었으며, 이러한 특성들에서 크게 벗어나는 화합물은 경구투여가 가능한 약물로 성공적이지 못하리라는 공감대가 형성되었다.[1] 따라서 이전과 같이 무작정 많은 화합물을 탐색하기보다는 화합물의 물리적인 특성에 먼저 관심을 기울여야 한다는 생각을 가지게 되었다. 실제로 약물의 친화도나 선택성을 올리기 위한 최적화 과정에서는 필연적으로 약물의 분자량이 커지게 되고 지질 친화도가 높아지는 경우가 많은데, 이 경우에는 결국 얻어지는 최종 산물이 리핀스키의 5규칙을 넘어서 경구투여 약물로 부적합해지는 경우가 많아지게 되었다. 따라서 이러한 문제에 직면하지 않으려면 처음에 얻어지는 히트의 분자량은 최종 약물로 사용되는 화합물에 비해 훨씬 작은 편이 유리할 것이다.
'프래그먼트 기반의 신약개발(Fragment based drug discovery)'는 기존의 HTS에 비해서 훨씬 더 분자량이 작은(150달톤에서 300달톤 이하의) 화합물 프래그먼트 라이브러리로부터 표적 단백질에 결합하는 화합물을 찾는데서 시작한다.[2] 물론 분자량이 작은 화합물은 일반적인 약물 후보물질에 비해서 훨씬 크기가 작고 단백질과 결합하는 부분도 작으므로 결합력은 약물과 비슷한 크기의 화합물에 비해서 훨씬 떨어지는 편이다. 그러나 보다 간단한 화합물을 이용하므로 분자량이 큰 화합물에 비해서 훨씬 적은 수인 1000개 이하의 화합물을 스크리닝해도 충분히 넓은 화학 다양성을 가진 화합물들의 결합 가능성을 테스트할 수 있다는 장점이 있다.
일반적으로 매우 낮은 결합력을 가진 프래그먼트의 저해 활성 측정은 기존의 HTS에서 사용하던 방법을 그대로 사용하기는 쉽지 않다. 따라서 NMR 분광법, 등온 적정 열량법(Isothermal Titration Calorimetry), 표면 플라스몬 공명법(Surface Plasmon Resonance, SPR), 마이크로스케일 열영동(Microscale Thermophoresis, MST) 등과 같은 물리적인 방법으로 표적 단백질과의 결합력을 측정하여 결합하는 작은 화합물인 프래그먼트(Fragment)를 찾게 된다.
이렇게 특정한 단백질에 결합하는 프래그먼트를 확보한 다음에는 이 프래그먼트와 표적 단백질과의 복합체의 결정 구조를 규명하게 된다. (아예 별다른 선별과정 없이 바로 대량의 단백질 결정화를 통하여 구조 규명에 의해서 단백질에 결합하는 프래그먼트를 선별할 수도 있다.) 이렇게 규명된 프래그먼트와 단백질간의 결합구조는 이후 선도물질을 개발하는데 이용된다. 단백질에 결합한 프래그먼트를 프래그먼트 결합 주변의 단백질 구조를 고려하여 좀 더 늘려서 단백질과 더 많은 상호작용을 할 수 있도록 만들어 결합력을 올릴 수도 있다. 만약 복수의 프래그먼트가 서로 인접부위에 결합한다면, 이를 서로 연결하여 결합력을 끌어올릴 수도 있다. 이렇게 만들어진 새로운 화합물과 단백질의 결합구조를 다시 결정하고 이의 저해 능력을 검정하는 과정을 반복하면서 물질을 최적화하여 발굴된 프래그먼트로부터 선도물질을 만들어 나간다.
이러한 프래그먼트 기반 신약개발은 필연적으로 화합물과 표적 단백질간의 결합구조 결정이 필수적이며, 이러한 구조정보에 의해서 지속적인 화합물 합성 및 단백질-화합물 복합체의 결정이 이루어지게 된다.
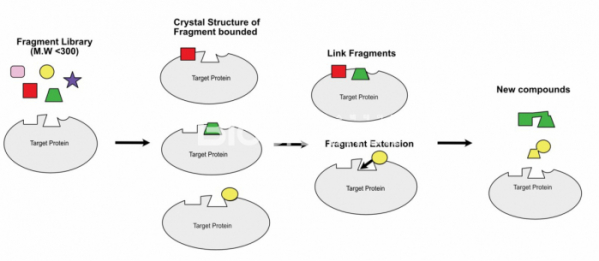
▲그림 1. 프래그먼트 기반 신약개발 과정의 개요. 분자량 300이하의 프래그먼트 라이브러리와 표적 단백질의 결정구조를 구한 후, 이에 기반하여 2개의 독립적인 위치에 결합한 프래그먼트를 서로 연결하거나, 혹은 프래그먼트 주변으로 화합물을 연장하여 더 나은 결합력을 가지는 화합물을 찾고, 이의 결정구조 결정을 반복하여 이 정보에 기반하여 화합물 개선을 계속 진행한다. 빈번한 단백질-화합물 결합구조의 결정은 프래그먼트 기반 신약개발에서 필수적인 요소가 된다.
가상 스크리닝
약물과 유사한 크기의 화합물이건, 혹은 약물보다 작은 프래그먼트가 되건, 수많은 화합물을 합성하고, 이의 생화학적/생물학적 활성을 측정하는데는 많은 비용과 시간이 든다. 그러나 이미 규명된 표적 단백질의 구조를 이용하여, 이 단백질 구조에 결합할 수 있는 화합물을 컴퓨터 상에서 탐색한다면, 실험에 비해서 훨씬 적은 비용으로, 훨씬 다양한 화합물을 테스트할 수 있을 것이다. 인실리코(In-sillico)에서 가상 화합물이 특정한 단백질에 어떻게 결합하는지를 알아보는 방법으로 도킹(Docking)이라는 방법이 있다. 도킹을 통하여 수백만, 수천만 종류의 가상 화합물 중에서 단백질의 활성자리에 결합할 만한 화합물을 추려내게 된다.
이렇게 도킹을 통해 얻을 수 있는 정보는 (1)특정한 단백질에 결합할 것으로 생각되는 화합물 (2) 가상 화합물이 단백질에 결합한 방식(포즈, Pose) (2)가상 화합물과 단백질과의 결합력 예측 등이다. 특히 가상 화합물이 단백질에 결합할 때의 단백질-소분자 상호작용이 기존에 단백질에 결합하는 것으로 알려지는 물질이 결합하는 방식과 유사하다면, 이렇게 예측된 결합 방식이 신빙성을 가질 가능성이 높다.
도킹에서 핵심이 되는 것은 화합물이 단백질에 결합된 형태를 평가하여 점수화하는 스코어링 펑션(Scoring Function)이다. 즉, 어떤 화합물이 단백질에 결합하는 정도를 수치화하여 보다 정확히 정량화할 수 있다면 수많은 화합물 중에서 좀 더 용이하게 선발할 수 있을 것이다. 도킹 스코어를 결정하는 방법을 대략적으로 분류하면 다음과 같이 분류된다.[3]
▲포스 필드 기반 스코어링 펑션(Force field based scoring function)= 화합물과 단백질간의 결합력을 이를 구성하는 원자간의 상호작용들의 물리적 성질에 기반하여 계산한다. 즉 화합물과 단백질 원자간의 여러가지 상호작용, 반데르발스 상호작용, 정전기 상호작용 등의 상호작용들을 기술하고 있는 포스 필드에 의해서 수치화하고 이를 합산하게 된다. 또한 화합물이나 단백질을 구성하고 있는 분자 내부의 에너지, 화합물의 용매화 역시 고려된다. 포스 필드 기반의 스코어링 펑션을 사용하는 도킹 소프트웨어로는 DOCK, GOLD 등이 있다.
▲경험 기반 스코어링 펑션(Empirical scoring function)= 실험적으로 측정한 결합력과 결합 복합체 구조가 알려져 있는 단백질-화합물의 데이터를 이용하여 만들어진 모델이다. 즉 화합물과 단백질 복합체 구조로부터 유래된 여러가지 상호작용에 관련된 수치와 결합력과의 관계를 통계적으로 분석하여 실험적인 결합력을 가장 잘 예측할 수 있는 요소들을 파악하고, 이들 요소와 결합력과의 관계를 다중회귀분석을 통하여 만들어지게 된다. 경험적 스코어링 펑션을 사용하는 도킹 시스템으로는 GLIDE, Autodock Vina, X-Score 등이 있다.
▲지식 기반(Knowledge-based)= 현재까지 규명되어 있는 단백질-화합물 결합구조 및 화합물 구조 데이터베이스에서 관찰된 원자간의 상호작용 분포로부터 평균 힘 전위(Potential of Mean Force)를 구하고 이를 이용하여 화합물과 단백질 도킹의 확률을 계산한다. 이러한 지식 기반의 스코어링 펑션에는 DrugScore가 있다.
그렇다면 실제로 화합물의 단백질의 결합을 예측하는 과정은 어떤 식으로 진행되는가? 일단 단백질 구조에서 결합을 탐색할 부분을 정한다. 물론 화합물이 단백질의 어디에 붙을지 모르는 상황에서 단백질의 전 영역에서 탐색할 수도 있지만, 이 경우에는 탐색의 시간도 길어지고 정확도도 떨어지기 때문에 단백질 구조를 참조하여 화합물이 결합할 만한 ‘포켓'이 있는 영역을 중심으로 결합을 탐색할 영역을 정한다. 이렇게 정해진 검색영역은 격자(grid) 형태로 나뉘게 되고, 이 영역 내에서 화합물의 결합 방식을 찾게 된다. 이를 위해서 화합물이 취할 수 있는 다양한 3차 구조를 형성한 다음 하고, 격자 내에 배열하게 된다. 이렇게 배열된 화합물과 단백질의 복합체에 대해서 스코어링 펑션을 이용하여 도킹 스코어를 계산하고, 가장 높은 스코어를 가지는 화합물의 포즈를 선택하게 된다.
이러한 단백질 도킹은 단백질에 결합하는 미지의 화합물을 찾아내는 방법으로 널리 이용되고 있지만, 가장 높은 도킹 스코어를 가지는 화합물이 그대로 가장 강한 결합력을 가지는 화합물로 검증되는 것은 아니다. 즉, 현재 개발된 도킹 소프트웨어와 스코어링 펑션은 수많은 화합물 중에서 단백질에 결합할 가능성이 있는 화합물의 비율을 추려내는 정도의 역할은 잘 할 수 있지만, 화합물의 결합력을 정확히 예측하는 방법은 아니기 때문이다. 화합물의 결합력을 정확하게 추정하려면 분자 동역학(Molecular Dynamics) 기반의 다른 방법을 사용해야 한다.
분자 동역학 기반의 화합물 검증방법
화합물 도킹을 위한 방법이 정확히 화합물과 단백질의 결합력을 예측하지 못하는 이유는 무엇일까? 여기에는 여러가지가 있겠지만, 도킹은 근본적으로 단백질을 고정된 수용체로 보고, 여기에 자유롭게 다른 형태를 가질 수 있는 화합물을 결합시키는 방법이라는데 한계가 있다. 실제의 상황은 단백질 역시 수용액 속에서 미세하게 움직임을 계속하고, 화합물과의 결합 형태도 계속 달라질 수 있다.
단백질과 화합물의 동적인 성격을 고려하려면 분자 동역학(Molecular Dynamics) 기반의 계산법이 필요하다. 분자 동역학은 단백질과 물분자, 그리고 단백질에 결합되어 있는 화합물 등 시스템을 구성하는 원자의 상호작용을 고전역학적인 방법으로 계산하여 단백질의 동적인 움직임을 알아보는 방법이다. 단백질-화합물 복합체를 분자 동역학으로 시뮬레이션하면, 단백질-화합물 복합체의 시간적인 움직임을 파악할 수 있으며, 이들의 상호작용이 얼마나 안정적인지, 시간에 따라서 어떻게 변하는지를 알 수 있게 된다. 화합물과 단백질이 얼마나 안정적으로 결합하고 있는지, 그리고 화합물에서 안정적으로 결합하는 부분과 그렇지 않은 부분이 어디인지에 대한 정보를 보다 정확히 할 수 있게 되므로 도킹 등의 방법에서 얻은 후보 화합물들의 특성을 좀 더 자세히 조사할 수 있다.
이러한 분자 동역학 방법에 기반하여 화합물과 단백질의 결합 자유 에너지를 비교적 정확히 예측할 수 있는 방법도 개발되었다. 자유 에너지 교란법(Free Energy Perturbation)이라고 불리는 방법으로써, 이 방법은 2개의 유사하지만 다소 상이한 화합물의 결합 자유 에너지를 분자 동역학 시뮬레이션을 통하여 계산하게 된다.[4] 이 방법은 화합물과 단백질에 따라서 다소 오차는 있지만 실험에 근거한 화합물의 결합 자유 에너지와 약 0.5-1 kcal/mol 내외의 오차로 예측 가능하다. 이 방법은 제약회사 등에서 화합물의 활성을 최적화하는 단계에서 다양한 화합물의 유도체를 만들고, 이들이 원 화합물보다 좀 더 결합력이 좋아졌는지, 그렇지 않은지를 미리 예측해 볼 때 널리 사용하게 된다. 이렇게 인 실리코 상에서 화합물의 결합력을 미리 예측하여 결합력이 상승된 것만 합성하여 실험을 함으로써, 실제 실험에 드는 비용을 절약할 수 있게 된다.
이러한 분자 동역학적인 화합물 검증 방법의 단점이라면 계산에 많은 시간이 소요되고, 따라서 대량의 화합물을 대상으로 계산을 돌리기는 어렵다는 데 있다. 최근의 컴퓨터 리소스의 발전, 특히 GPU에 의한 고속 연산이 가능해져서 조금 일반화되긴 했지만, 오늘날의 현대적인 컴퓨터에서도 하나의 단백질-화합물 복합체의 분자동역학 시뮬레이션을 수행하기 위해서는 최소 몇 시간에서 수십 시간의 시간이 걸린다. 따라서 분자 동역학 기반의 계산방법은 가상 스크리닝의 첫 단계에서는 사용하기 힘들고, 도킹 등의 방법으로 일차적으로 후보군을 선별한 후 남은 소수의 화합물에 대해서 사용하는 것이 보통이다.
화합물과 단백질 구조를 고해상도로 풀게 되면 가끔 화합물이 결합한 옆에 물 분자가 같이 위치하고 있는 경우가 있다. 결정 속에는 물이 항상 존재하지만 결정 구조에서 물 분자가 관찰된다는 것은 결정을 구성하고 있는 모든 구성물에서 항상 같은 위치에 물이 결합되어 있다는 의미라는 점에서 중요하다. 즉, 화합물의 결합에 물 분자 역시 중요한 역할을 하는 것이다.
이렇게 화합물과 단백질에 안정적으로 결합하고 있는 물 분자는 분자 동역학 시뮬레이션을 통해서도 확인 가능하다. 분자 동역학 시뮬레이션을 통하여 추가적으로 얻을 정보는 각각의 물 분자가 어떤 에너지 상태에 있는가이다. 가령 물 분자 중에서 화합물과 단백질의 결합에 긍정적인 역할을 하는 것도 있는 반면, 어떤 경우에는 물 분자가 없는 쪽이 화합물의 결합에 도움이 되는 것도 있다. 단백질 결정학을 통하여 파악한 화합물 근처의 물 분자의 에너지 상태를 분자 동역학 시뮬레이션으로 확인하고, 그 중 불안정한 물 분자가 있는 위치까지 화합물이 차지하도록 새롭게 디자인하게 되면 화합물의 결합 활성이 높아지는 경우가 많이 있었다.[6] 이러한 기법은 현재 구조 기반으로 화합물을 최적화할 때 일반적으로 사용되는 방법론이 되었다.
버츄얼 스크리닝 깔때기(Virtual Screening Funnel)
이러한 여러가지 구조 기반의 화합물 버추얼 스크리닝 방법은 현대의 신약개발에서 필수적으로 사용되는 테크닉이 되었다.
통상적인 버추얼 스크리닝의 경우 일단 Zinc(https://zinc20.docking.org), PubChem (https://pubchem.ncbi.nlm.nih.gov)에서 얻을 수 있는 화합물의 데이터로부터 시작한다. 이러한 화합물 중에서 분자의 여러 특성을 고려하여 약물과 비슷한 성질을 가지는 화합물을 추려내고, 범 분섭 간섭 화합물(pan-assay interference compounds, PAIN)과 같이 화합물 어세이에서 위양성을 주는 화합물들을 걸러내는 것으로 시작한다. 이렇게 걸러낸 화합물에서 후보물질을 추출하는 것에는 도킹 등의 구조기반 방법과 화합물의 구조에 기반한 방법 둘로 나뉜다. 도킹 등의 구조기반 방법에서는 일반적으로 일차 스크리닝 용으로는 빠르게 많은 화합물을 검색할 수 있는 조건으로 수천만개가 넘는 화합물을 검색한 후, 상위 1-10% 화합물에 대해서 좀 더 정밀한 도킹이 가능한 스코어링 펑션 혹은 방법을 이용하여 좀 더 정확한 스코어를 얻는다. 만약 해당하는 단백질에 이미 결합하는 화합물 구조가 알려져 있는 경우, 비슷한 방식으로 결합하는 화합물을 선발하기도 한다. 화합물에 기반한 방법은 특정한 단백질에 작용하는 약물작용단(Pharmacophore)이 알려져 있는 경우, 이러한 약물작용단을 가지고 있는 화합물을 데이터베이스에 검색하여 유사 화합물을 검색하는 것으로 시작된다. 이러한 구조 기반 방법과 화합물 기반의 방법은 병행되거나 같이 접목되어 사용되기도 한다. 최종적으로 실험적으로 검증을 거치기 전에 골라낸 비교적 소수의 화합물(수백개 이내)에 대하여 자유 에너지 교란법과 같이 비교적 많은 계산이 요구되는 방법이 사용되기도 한다. 이렇게 버추얼 스크리닝에서 다단계의 방법으로 수많은 화합물로부터 시작하여 실험적 검증이 가능한 수준의 소수의 후보 화합물로 줄여나가는 과정을 ‘버추얼 스크리닝 깔대기(Virtual Screening Funnel)'이라고 칭하기도 한다.
이러한 버추얼 스크리닝은 특히 코로나19 팬데믹과 같이 빠르게 후보물질 도출이 요구되는 상황에서 중요한 역할을 하게 되었다. 2022년 스웨덴의 웁살라 대학 연구팀에서 SARS-CoV-2 바이러스의 주 단백질 분해효소의 활성을 저해하는 화합물을 스크리닝하는 과정을 살펴보면 ‘버추얼 스크리닝 깔때기’의 역할을 알 수 있다. 이들은 약 2억3500만개의 화합물을 SARS-CoV-2 주 단백질 분해효소에 도킹하였고, 여기서 상위 30만개의 화합물을 화합물의 유사성으로 분류한 후, 기존에 알려져 있는 저해물질과 결합 방식이 동일한 화합물을 찾아서 약 82개의 화합물을 실험 검증 대상으로 선별하였다. 여기서 선별된 화합물 중 가장 결합 활성이 좋았던 화합물을 대상으로 여러 단계의 구조 기반 화합물 최적화 작업을 실시하여, 최종적으로 현재 사용되고 있는 단백질 분해효소 저해제와 유사한 수준의 활성을 얻을 수 있었다.[6] 이러한 예는 오늘날 구조 기반의 버추얼 스크리닝이 소분자 기반의 신약개발에 어떻게 활용되는지를 잘 보여주고 있는 셈이다.
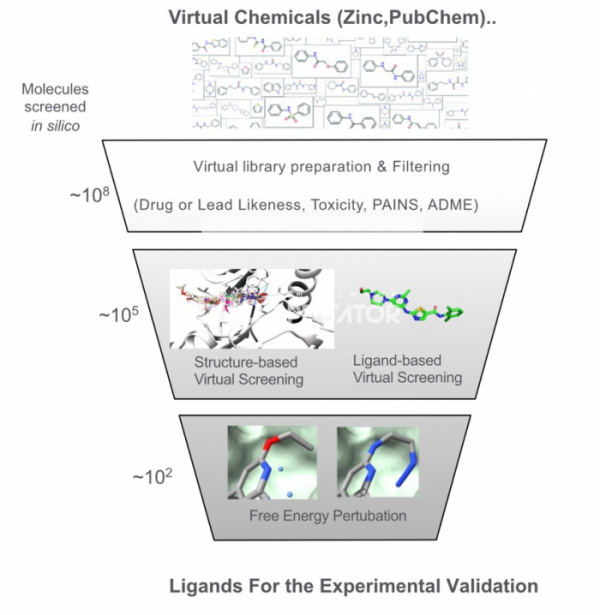
▲그림 2. 버츄얼 스크리닝 깔때기(Virtual Screening Funnel) : 조합화학의 발전으로 현재 수억 종류의 화합물이 상업적인 벤더로부터 쉽게 입수할 수 있게 되었고, 이러한 가상 화합물 정보를 이용하여 표적 단백질에 결합하는 후보 화합물을 선별하는 버츄얼 스크리닝은 오늘날 신약 개발에 널리 사용되고 있다. 버츄얼 스크리닝은 여러 단계를 거쳐서 최종적으로 실험 검증에 들어갈 수백개 이내의 화합물을 선별하며, 이러한 과정을 버츄얼 스크리닝 깔때기(Virtual Screening Funnel)라고 칭한다.
참고문헌
1. Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews, 23(1-3), 3-25.
2. Murray, C. W., & Rees, D. C. (2009). The rise of fragment-based drug discovery. Nature chemistry, 1(3), 187-192.
3. Liu, J., & Wang, R. (2015). Classification of current scoring functions. Journal of chemical information and modeling, 55(3), 475-482.
4. Wang, L., Wu, Y., Deng, Y., Kim, B., Pierce, L., Krilov, G., ... & Abel, R. (2015). Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. Journal of the American Chemical Society, 137(7), 2695-2703
5. Abel, R., Young, T., Farid, R., Berne, B. J., & Friesner, R. A. (2008). Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. Journal of the American Chemical Society, 130(9), 2817-2831.
6. Luttens, A., Gullberg, H., Abdurakhmanov, E., Vo, D. D., Akaberi, D., Talibov, V. O., ... & Carlsson, J. (2022). Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. Journal of the American Chemical Society, 144(7), 2905-2920.