기사본문
[남궁석의 신약연구史]단백질 구조 기반의 신약개발
입력 2022-06-07 09:34 수정 2022-06-13 10:09
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
지난 연재에서 암, 바이러스 등의 질병관련 단백질 구조가 밝혀지는 과정을 살펴보았다. 결국 질병관련의 기전과 단백질 구조를 연구하는 궁극적인 목적은 질병을 치료할 수 있는 방법을 찾기 위한 것이다. 그렇다면 어떻게 질병관련 단백질의 구조가 해당 질병을 치료하는 약물의 개발로 이어질 수 있을까? 흔히 신약개발 과정에 정통하지 않은 많은 사람들(심지어 많은 생물학자 역시)은 질병관련 단백질의 구조가 규명되면 이러한 단백질을 억제하는 약물이 탄생하는 것은 시간문제라고 생각한다.
실제로 단백질의 구조에 대한 정보는 이 단백질에 결합하는 화합물을 단백질의 작용원리에 따라서 찾을 수 있게 하는 매우 중요한 정보다. 그러나 어떤 단백질에 강하게 결합하여 이 단백질의 기능을 저해하는 물질을 찾는 것은 질병을 치료하는 약물을 개발하는 과정에서 가장 처음의 단계에 불과하다. 시험관 밖의 실험에서 표적단백질에 강하게 결합하여 그 활성을 저해하는 물질을 찾았다고 하더라도, 이 화합물이 반드시 우리 몸 속에서 약으로 작용하여 병을 치료한다는 보장은 없다. 약물이 흡수가 제대로 되지 않아 원하는 세포 내에 전달이 잘 되지 않을 수도 있으며, 약물이 우리 몸 속에서 너무 신속하게 분해되어 약효를 내기도 전에 사라져 버릴 수도 있다. 또한 우리가 목적하는 타깃 이외의 단백질에 작용하여 심한 독성을 낼 수도 있다.
이러한 여러가지 요인들을 전임상 연구단계에서 테스트하고 인간 대상의 임상시험에 들어가도, 정작 기대한 만큼의 효과를 내지 못하거나 부작용이 심해 약물로써의 가치가 없는 것으로 판명되는 경우가 훨씬 더 많다. 이렇게 신약개발은 다단계의 조건을 최적화해야 하는 문제로, 단순히 특정한 표적단백질의 구조를 아는것은 신약개발에 필요한 여러가지 정보 중 하나일 뿐이다. 그리고 표적단백질의 구조를 알아도 이 단백질에 약물이 강하게 결합할 수 있는 결합자리(Binding Pocket)이 없다면 이 표적단백질의 기능을 저해하는 물질 자체를 찾는 것이 어려울 수도 있다.
이러한 여러가지 어려움 때문에 질병관련 단백질의 구조가 알려지고 나서도 이러한 구조를 통해 개발된 신약의 갯수는 생각만큼 많지 않다. 그러나 이러한 어려가지 어려움을 딛고서 그간 여러 종류의 약물이 개발되었으며, 단백질 구조 기반의 신약개발은 적어도 소분자 화합물 기반 신약개발에 있어서 기본적인 과정이 되었다. 앞으로 2회의 연재를 통하여 어떻게 단백질 구조 기반의 신약개발이 소분자 화합물 기반의 신약개발 과정의 기본이 되어가는지를 알아보도록 한다.
바이러스 질병관련 약물의 구조 기반 개발
단백질 구조 정보가 약물의 개발에 실질적인 보탬을 준 약물의 첫번째 예는 바이러스 질병에 관련된 약물, 특히 HIV 치료제였다. 지난 연재에서 HIV의 단백질 분해효소가 HIV의 증식 과정에서 매우 중요한 역할을 한다는 것을 설명하였다. 따라서 HIV 단백질 분해효소를 억제하면 HIV 증식을 억제하여 에이즈를 치료할 수 있다는 가정하에 많은 연구자 및 제약회사들이 HIV 단백질 분해효소를 억제할 수 있는 물질을 만들고자 하였다. 그렇다면 어떻게 HIV 단백질 분해효소를 억제할 수 있을까?
많은 연구자들은 실제 HIV 단백질 분해효소가 절단하는 HIV의 단백질 서열을 분석하고, 이와 유사하지만 이를 약간 변형한 펩타이드 유사체를 이용하면 HIV 분해효소에 결합하여 이를 억제하는 억제물질을 만들 것이라고 생각했다. 그러나 이 과정을 최적하려면 일단 HIV 분해효소가 어떻게 단백질(혹은 억제물질)과 결합하는지에 대한 정확한 정보가 있어야만 했다. 1989년 미국 암연구소(National Cancer Institute)와 캘리포니아공대(Caltech)의 공동연구팀은 HIV의 단백질 분해효소와 이 단백질 분해효소가 인식하는 6개의 아미노산으로 이루어진 서열 내에서 펩타이드 결합을 형성하는 카르복시기를 변형하여 단백질 분해를 일으키지 못하게 한 유도체와의 결합구조를 밝혔다. 아스파르트산 단백질 분해효소의 촉매과정에서 핵심적인 역할을 하는 아미노산인 25번째 아스파르트산은 HIV의 단백질 분해효소에 의해서 분해되는 부분(실제 구조에서는 변형되어 분해 반응이 일어나지 않지만)과 근접한 위치에 위치하고 있었다.[1]
그러나 단순히 단백질 분해효소가 자르는 펩타이드 서열을 약간 변형하는 것만으로는 약물로 쓸 만큼 강력한 저해활성을 얻기 힘들었고, 펩타이드는 세포내로의 흡수가 잘 되지 않으며 체내에 존재하는 단백질 분해효소에 의해서 빠르게 분해되기 때문에 약물로 바로 사용할 수는 없었다. 이러한 단점을 극복하기 위해서, 실제의 단백질 분해효소에 의해 분해되는 펩타이드와 거의 유사하게 결합되지만 약물로의 여러가지 특성이 우수한 화합물을 개발해야만 했다.
다국적 제약회사 로슈(Roche)의 연구자들은 HIV 단백질 분해효소가 HIV 단백질 중 페닐알라닌-프롤린, 혹은 타이로신-프롤린의 사이를 절단한다는 것에 주목했다. 동물의 일반적인 아스파르트산 단백질 분해효소는 이러한 아미노산 사이를 절단하는 경우가 거의 없기 때문에, 만약 이러한 아미노산을 특이적으로 인식하는 단백질 분해효소를 저해하는 물질을 만든다면 숙주세포의 단백질 분해효소를 그닥 저해하지 않고 바이러스의 단백질 분해효소만을 특이적으로 저해하는(따라서 별다른 부작용이 없는) 물질을 만들 수 있을 것이라고 가정하였다. 이들은 페닐알라닌과 프롤린으로 이루어진 다이펩타이드를 단백질 분해효소에 의해서 분해되지 않도록 변형한 하이독시에틸아민(hydroxyethylamine) 유도체를 만들었고, 이 물질은 HIV 단백질 분해효소에 약한 저해활성을 가졌다. 이렇게 처음 찾은 페닐알라닌과 프롤린 다이펩타이드 기반의 화합물을 연장해 가며 약 100여개의 유도체를 만들었고 이 중 한 화합물은 0.4나노몰(nM IC50)이하의 강한 저해활성을 가지고 있었다.[2] 이 물질과 HIV 단백질 분해효소의 결합구조를 결정해 보았더니, HIV 단백질 분해효소의 단백질 분해에 핵심적인 잔기인 25번째 아스파르트산이 마치 천연 단백질과 결합한 상태와 유사하게 결합하고 있었지만, 화합물의 구조는 천연 단백질의 구조와 미세하게 다르기 때문에 화합물은 분해되지 않고, 효소활성은 저해되게 된다.[3]
이렇게 개발된 화합물은 ‘사퀴나비르(Saquinavir)'라는 정식 물질명이 되고 1995년 HIV 단백질 분해효소 저해제로 최초로 등장한 약물이 되었고, 사퀴나비르의 뒤를 이어 여러 종류의 HIV 단백질 분해효소 저해제가 등장했다.
이렇게 개발된 HIV 단백질 분해효소 저해제는 다른 항바이러스 물질과 조합하여 ‘칵테일 요법' 이라고 불리는 ‘고활성 항레트로바이러스 요법(Highly active antiretroviral therapy, HAART)'의 핵심 구성요소가 되었다. 돌연변이가 빠른 HIV에 의해 유발되는 에이즈는 하나의 항바이러스 물질에만 의존하면 금방 이 물질에 내성을 가지는 변이체가 나타난다. 그러나 동시에 2개 혹은 3개의 별도의 기전을 가진 약물을 사용하는 HAART는 상대적으로 치료법에 내성을 가지는 바이러스가 나타나기 어려웠으며, 따라서 HIV 감염자에 대한 좋은 치료법이 되었다. 1990년대 중반 도입된 HAART가 HIV 감염자에 미친 영향은 지대하다. HAART 도입 이전인 1989년 경 HIV에 감염된 20세 청년의 평균 기대 여명은 11.9년이었다. 즉 그 당시의 치료법의 경우 HIV에 감염된 20세 청년은 평균적으로 30세 중반이 되기 전에 숨진다는 이야기다. 그러나 HAART가 일반화된 이후인 2006-2013년 20세 HIV 감염자의 기대 여명은 54.6년이었다. 고학력·고소득자와 같이 HAART에 의한 치료를 더 잘 받고 건강관리가 잘 될 것으로 기대되는 계층의 경우 HIV 감염자의 기대 여명은 60년으로, 일반 대중과 별 차이가 없었다. 결국 HIV 단백질 분해효소 저해제를 통해 열린 HAART는 에이즈를 불치의 질병으로부터 지속적으로 치료만 잘 받는다면 오랫동안 건강을 유지하면서 살 수 있는 당뇨나 고혈압과 같은 만성질병으로 바꾸어 놓은 셈이다.[4]
HIV 단백질 분해효소 저해제의 개발로 시작된 단백질 구조 정보를 기반으로 하는 항바이러스제의 개발은 그 이후에도 계속 이어졌다. 인플루엔자 치료제로 유명한 타미플루는 인플루엔자 바이러스의 감염에 필수적인 효소인 뉴라미데이즈(Neuramidase)에 결합하는 물질로, 이의 개발에도 뉴라미데이즈의 구조 및 저해물질의 결합구조가 중요한 역할을 하였다.
최근의 코로나바이러스 팬데믹에서도 구조기반의 신약개발은 큰 효과를 발휘했다. 코로나바이러스 역시 HIV 등과 마찬가지로 바이러스를 구성하는 단백질은 일단 한 가닥의 단백질로 만들어지고, 이것이 바이러스가 가진 단백질 분해효소에 의해서 절단되어 코로나바이러스를 구성하는 각각의 단백질이 된다. 2019년 SARS-CoV-2 팬데믹 이후 수많은 제약사들은 SARS-CoV-2 의 저해활성을 가지는 물질을 개발하려고 하였으며, 코로나바이러스의 주 단백질 분해효소(Main Protease)는 주된 표적이 되었다. 사실 2002년의 사스 코로나바이러스가 처음 출현하였을때 화이자(Pfizer) 등의 제약사들은 바이러스 치료제를 만들기 위해 코로나바이러스의 주 단백질 분해효소를 타깃으로 하여 후보물질을 찾아두었다. 그러나 사스 코로나바이러스가 다시 출현하지 않고 사라지지 않자 찾은 선도물질은 더이상 개발되지 않고 사장되어 있었다. 그러나 코로나19 팬데믹 이후 신속한 바이러스 치료제를 찾으려는 시도에 의해서 이 물질은 다시 빛을 보게 되었다.
원래의 선도물질은 바이러스 저해활성은 강했으나 경구투여했을때 거의 흡수되지 않아 먹는 약으로 사용할 수 없는 상태였다. 그러나 팬데믹 상황에서 매일 복용해야 하는 약물의 경우 주사제보다는 먹는 약으로 제조할 필요가 있고, 이를 위해 경구투여가 가능하면서도 바이러스 저해 활성을 그대로 유지한 물질을 만들어야만 했다. 이를 위해 여러가지 다양한 물질과 주 단백질 분해효소간의 복합체 구조가 결정되었으며, 이러한 단백질 구조에서 유추된 결합방식에 근거하여 새로운 물질이 디자인되었으며, 얻어진 물질은 코로나바이러스의 주 단백질 분해효소의 촉매작용에 필수적인 역할을 하는 145번 시스테인과 공유결합을 할 수 있는 새로운 물질이었다.[5] 이렇게 얻어진 니르마트렐비르(Nirmatrelvir)라는 물질은 체내에서의 지속시간을 늘리는 리토나비르(ritonavir)로 함께 임상과 임상시험을 거쳐서 2021년 말 ‘팍스로비드(Paxlovid)'라는 이름의 치료제로 널리 공급되기 시작하였다. 불과 2년 이내에 전혀 새로운 화학구조를 가진 새로운 치료물질을 개발하는데 구조생물학의 역할은 매우 중요했다. 이렇게 구조생물학에 의한 신약개발에서 가장 먼저 활발히 이루어진 것은 HIV나 인플루엔자 바이러스 등의 바이러스 질병 치료제였다. 그러나 제약업체들의 신약개발에서 가장 높은 비중을 차지하고 있는 항암 신약개발에서도 점차 구조생물학은 중요한 역할을 하기 시작했다.
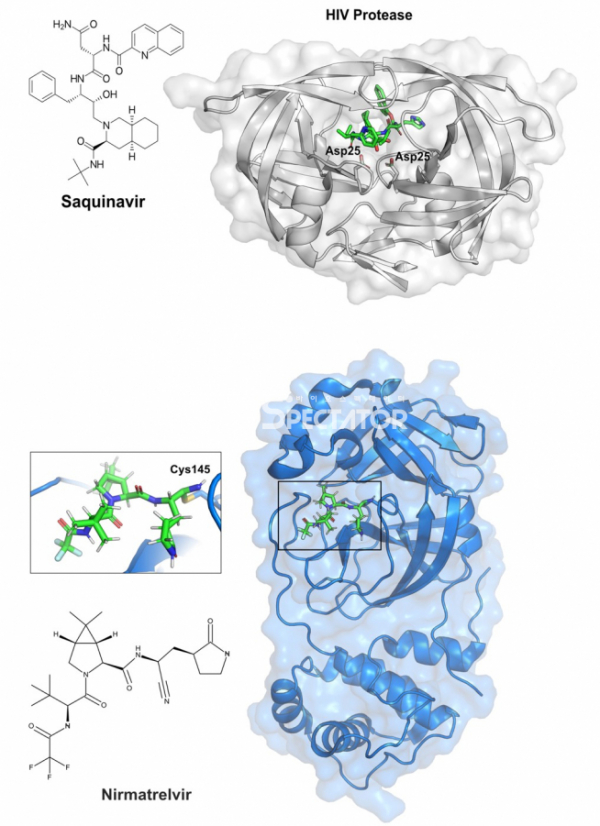
▲그림 1. 바이러스 단백질 분해효소와 여기에 결합하는 저해제 (상) HIV 단백질 분해효소와 여기에 결합한 저해물질인 사퀴나비르. 아스파르트산 단백질 분해효소인 HIV 단백질 분해효소의 핵심 아미노산인 25번째 아스파르트산(Asp25)는 사퀴나비르와 인접하여 위치해 있다. (하) SARS-CoV-2 주 단백질 분해효소에 결합한 니르마트렐비르(Nirmatrelvir). 니르마트렐비르는 시스테인 단백질 분해효소인 코로나바이러스 주 단백질 분해효소의 145번째 시스테인(Cys145)와 공유결합을 형성하여 단백질의 활성을 억제한다.
단백질 인산화 효소 저해제와 단백질 구조
지난 회에서 설명한 것처럼 1980년대 중반에 많은 암 유전자가 돌연변이가 발생한 단백질 인산화 효소라는 것이 알려진 이후, 많은 연구자들은 단백질 인산화 효소를 저해하는 물질을 항암 표적치료제로 사용할 수 있을 것이라고 생각하고 이러한 물질을 찾으려고 노력했다. 그러나 단백질 인산화효소가 유효한 항암 표적이 되기 어렵다고 생각하는 회의적인 의견도 많았는데, 그 이유는 우리 몸 속에는 약 500종에 달하는 단백질 인산화효소가 있고, 이들 거의 대부분은 동일한 인산화 도메인을 공유하고 있다는 것이다. 단백질 인산화효소의 인산화 도메인에 결합하여 단백질 인산화효소를 저해하는 물질은 다른 단백질 인산화효소를 저해할 가능성도 많고, 따라서 원하지 않는 독성이 나타날 가능성도 많다는 주장이었다. 실제로 단백질 인산화효소 연구 초창기에 알려진 타이로신 유사체 기반의 저해물질들은 이렇다할 선택성을 가지지 않고 단백질 인산화효소는 모두 저해하는 것으로 나타나서 이러한 우려는 어느정도 근거가 있는 셈이었다.
그러나 특정한 단백질 인산화효소에 특이적으로 작용하는 화합물이 탄생함으로써 실제로 단백질 인산화효소를 타깃으로 하는 항암제가 개발될 수 있다는 것이 입증되게 되었다. 지난 회에 잠시 알아보았던 만성 골수성 백혈병(chronic myeloid leukemia)는 9번 염색체와 22번 염색체의 융합으로 생기는 BCR-ABL 융합 유전자에서 생성되는 조절기능이 망가진 BCR-ABL 단백질 인산화효소에 의해 일어나는데, 스위스 제약사인 시바-가이기(현 노바티스)는 1997년 ABL 단백질 인산효소를 특이적으로 저해하는 STI-571이라는 물질을 개발하였고, 이 물질은 ABL의 경우 약 38nM의 저해 높은 저해활성을 가지고 있었지만 EGFR 인산화효소, c-Src, PKA 등의 인산화효소들은 거의 저해하지 않는 높은 특이성을 가지고 있었다.[6] 이 물질은 곧 만성 골수성 백혈병 치료에 탁월한 효과를 가지고 있다는 것이 입증되었고, 1999년 이마니티브(Imanitib)라는 정식 성분명으로 판매가 허가되어 ‘글리벡(Gleevec)'이라는 제품명으로 알려지게 되었다.
그렇다면 이러한 이마니티브의 높은 선택성은 어떻게 가능한 것일까? 이마니티브가 처음 개발될 때는 ABL과 이마니티브의 결합구조가 알려지지 않은 상태였고, 연구자들은 이마니티브가 지난 회에 소개한 단백질 인산화효소의 활성화된 형태에 결합하고 있는 것으로 추측하였다. 그러나 2000년 미국 캘리포니아 대학의 존 쿠리안 연구실에서 이마니티브가 ABL에 결합되어 있는 구조를 규명하였고, 원래의 예상과는 달리 이마니티브는 단백질 인산화효소의 활성화된 상태가 아닌, 단백질 인산화효소의 불활성화된 상태에 결합하고 있다는 것을 알게 되었다.[7] 즉 이마니티브는 활성화된 단백질 인산화효소에 결합하여 단백질 인산화를 못하게 하는 것이 아니라, 단백질 인산화효소의 불활성화된 상태에 결합하여 단백질 인산화효소가 활성화된 상태로 변화하는 것을 막아 줌으로써 역할하는 것이었다.
이러한 독특한 결합방식은 왜 이마나티브가 ABL에 특이적인 저해물질이 되었는지를 잘 설명해 준다. 앞에서 이야기한 것처럼 단백질 인산화효소는 대부분 유사한 인산화 도메인을 공유하고 있고, 이들의 형태는 특히 활성화된 상태에서 유사하다. 그러나 활성화된 단백질 인산화 효소에 비해서 활성화 루프가 닫힌 형태의 단백질 인산화효소는 단백질 인산화효소에 따라 구조의 차이가 크고, 이러한 구조의 차이에 의해서 이마니티브는 ABL 인산화효소에 특이적인 약물이 될 수 있었다.
이후에 다양한 단백질 인산화효소를 대상으로 2020년까지 약 50여종의 단백질 인산화효소 저해제가 FDA로부터 허가 승인받았으며, 이러한 많은 종류의 단백질 인산화효소의 개발 과정에서 단백질 구조의 정보는 필수적인 역할을 했다. 때로는 단백질 구조가 단백질 인산화효소의 돌연변이에 의한 약물의 내성을 극복하는 약물을 만드는데 중요한 역할을 하기도 한다. 이마니티브가 사용되기 시작한지 오래 지나지 않아, 이마니티브 투여를 받은 환자에게서 이마니티브에 내성을 가진 암세포가 증식한다는 것이 밝혀졌다. 이의 발생기전을 살펴본 결과 ABL 인산화효소의 315번째 쓰레오닌이 이소류신으로 바뀐 돌연변이(T315I)가 이마니티브의 결합을 방해하여 내성이 생기는 것으로 확인되었다.[8]
이러한 내성을 어떻게 극복할 수 있을까? 바이오텍인 아리아드 파마슈티컬(ARIAD Pharmaceutical)은 일본 제약사 오츠카제약과 합력하여 T315I 돌연변이를 가진 ABL 인산화효소에도 결합하여 저해할 수 있는 저해제인 포나티닙을 개발했다.[9] 포나니팁과 이마니팁, 그리고 ABL 인산화효소의 결합 구조를 보면 왜 이마니팁은 T315I 돌연변이에 취약하지만 포나니팁은 돌연변이의 존재에 크게 영향받는지를 쉽게 알 수 있다. 315번 쓰레오닌의 히드록시(OH)기는 이마니팁에 있는 아미노기의 질소 원자와 가까이 위치하고 있으며 서로 상호작용을 할 수 있다. 그러나 쓰레오닌이 소수성 아미노산인 이소류신으로 바뀌게 되면 이소류신의 소수성 잔기와 아미노기간의 상호작용에는 문제가 생긴다. 그러나 포나니팁은 이 위치에 탄소 간의 3중결합이 존재하고 있고, 소수성을 띈 탄소-탄소 삼중결합은 이소류신과 상호작용하는데 문제가 없으므로 결합에 문제가 없게 된다. 이렇게 내성 돌연변이에 대응하는 포나니팁과 같은 화합물은 약물과 표적단백질간의 결합구조가 존재하지 않았다면 개발하는 것이 불가능에 가까웠을 것이다.
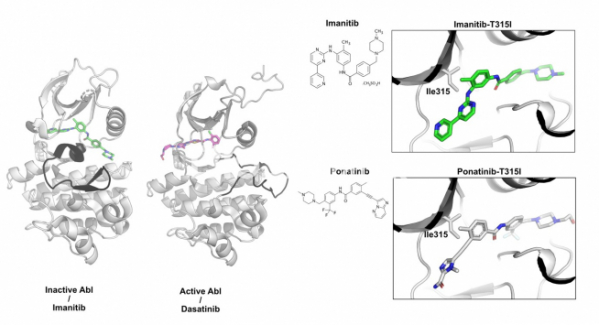
▲그림 2 . (좌)최초의 단백질 인산화효소 표적 치료제인 이마니티브와 Abl 인산화효소와의 결합 구조. 이마니티브는 ABL 인산화효소의 불활성화된 상태에 결합하여 불활성화된 상태를 그대로 유지시켜 주는 방식으로 단백질 인산화 효소 활성을 억제한다. 반면 일부 단백질 인산화효소 저해제(다사티니브 Dasatinib)는 활성화된 상태의 단백질 인산화효소에 결합하여 활성을 저해하는 경우도 있다. ABL 인산화효소의 활성화 루프(검은색으로 표시)는 이마니티브와 다사니티브의 결합 상태에 따라서 다르다. (우)이마니티브와 포나니티브의 결합 구조. 이마니티브는 ABL 인산화효소의 315번 쓰레오닌이 이소류신으로 바뀐(Ile315) 경우 결합력이 떨어진다. 이를 극복하기 위하여 새롭게 디자인된 저해제인 포나니팁은 315번 이소류신의 경우에도 정상적으로 결합하여 이마니티브 내성 돌연변이에도 작용할 수 있다.
K-ras 저해물질의 개발
이렇게 단백질 구조에 기반한 소분자 물질의 개발이 일반화되고 다양한 질병의 표적단백질을 저해하는 물질이 개발되고 있던 와중에도 유독 지지부진했었던 것은 이전 회에서 소개했던 K-ras 를 저해하는 저해물질이었다. 가장 일찍 알려진 암 유발 유전자였고, 가장 많은 종류의 암에서 돌연변이가 발생하는 K-ras를 표적으로 하는 약물은 왜 쉽게 등장하기 어려웠을까? 이는 지난 연재에서 소개한 Ras의 구조를 보면 어느정도 이해가 가능하다. 약물이 단백질에 표적으로 작용하기 위해서는 단백질의 가운데에 약물이 단단히 결합할 수 있는 움푹한 ‘포켓(Pocket)'이 있어야 한다. 그러나 상대적으로 작고 둥근 형태의 구조를 가지고 있는 K-ras에는 약물이 강하게 붙을 수 있는 깊은 포켓이 발견되지 않았고, 따라서 K-ras에 직접 결합하여 활성을 억제하는 화합물을 개발하는 것은 불가능할 것이라고 생각하였다.
따라서 K-ras를 타깃으로 하는 치료법을 개발하려고 하던 연구자들은 K-ras에 직접 결합하는 화합물보다는 K-ras의 특성을 이용하여 이의 활성화를 억제하는 방법을 만들려고 했다. K-ras 류의 단백질의 특성이라면 신호전달을 하기 위해서는 일단 단백질의 끝에 파네실화(Farnesylation)라는 반응에 의해서 지질이 결합하고, 이를 통하여 단백질이 실제로 작동하는 곳인 생체막에 결합하게 된다. 만약 K-ras에 파레실화가 일어나지 않게 된다면 K-ras는 실제로 기능을 수행하는 생체막으로 이동할 수 없기 때문에 작동하지 못하게 된다. 많은 제약회사들은 K-ras의 끝에 파레실화를 시키는 단백질인 파네실기 전이효소(Farnesyl transferastion)를 억제하면 K-ras가 세포 내에서 제 위치에 이동할 수 없으므로 기능을 억제할 수 있을 것이라고 생각하고 파레실기 전이효소를 억제하는 화합물을 개발하였다. 이렇게 개발된 화합물은 세포수준 및 실험동물 수준의 연구에서는 탁월한 항암효과를 보여 인간 대상의 임상시험으로 들어갔으나, 인간 대상의 임상에서는 전혀 항암 효과를 내지 못했다. 무엇이 문제였을까? 인간의 K-ras에서는 파레실화가 저해되더라도 다른 단백질 변형인 제라닐제라닐화가 일어나고 K-ras는 정상적으로 생체막으로 운반될 수 있었다. 결국 파레실기 전이효소를 억제하여 K-ras를 억제하려는 시도는 이를 개발하려는 많은 노력과 비용에도 불구하고 성공하지 못했다.[10]
이렇게 ‘공략 불가능’한 타깃이라고 여겨지던 K-ras의 저해물질을 개발하는데 돌파구가 열린 것은 2010년대 초반이었다. UCSF의 화학생물학 연구자인 케반 쇼캇(Kevan Shokat)은 K-ras에 분포하는 암 유발 돌연변이 중 G12C 돌연변이에 특이적으로 반응하는 화합물을 찾을 수 있다고 생각하였다. 즉 시스테인의 씨올(thiol, -SH)기에 공유결합으로 반응하는 화합물을 이용하면 K-ras에 특이적으로 결합하고 활성을 저해하는 화합물을 만들 수 있다는 아이디어였다. 쇼캇 연구실에서는 씨올 기에 반응할 수 있는 화합물 라이브러리를 이용하여 K-ras 활성을 억제하는 화합물을 찾았고, 이렇게 발견된 화합물과 K-ras의 결합구조를 규명해 보니, 화합물은 예상대로 12번째 시스테인에 공유결합으로 연결되어 있었고, 화합물이 결합되지 않은 단백질의 구조가 미세하게 변화하여 화합물이 강하게 결합할 수 있는 ‘포켓'이 형성되어 있고 여기에 화합물이 결합되어 있었다.[11]
이렇게 발견된 화합물은 당장 약물로 쓸 수 있을만큼 높은 활성을 가지고 있지는 않았다. 그러나 이러한 발견에 자극받은 제약회사들은 이와 비슷한 전략을 이용하여 G12C 돌연변이를 가진 K-ras를 저해하는 화합물을 찾기 시작했다. 이러한 회사 중 가장 먼저 성과를 올린 것은 암젠(Amgen)으로, 암젠은 2021년 K-ras G12C 돌연변이에 특이적으로 공유결합을 형성하여 저해하는 약물인 소토라십을 발굴하고[12] G12C K-ras 돌연변이를 가진 비소세포폐암 환자 대상으로 사용할 수 있는 허가를 취득하고 최초의 K-ras 표적 치료제로 등장하였다. 이후 미라티 세라퓨틱스(Mirati Therapeutics)가 G12C K-ras를 저해하는 다른 약물인 아다그라십(Adagrasib)이 2022년 사용 승인을 얻게 되었다.[13] 돌연변이 K-ras가 암을 유발하는 유전자임이 알려진지 40년이 지난 이후였다.
그렇다면 G12C가 아닌 K-ras의 돌연변이는 어떨까? 암을 유발하는 K-ras 돌연변이는 G12C 이외에도 G12D, G12V, Q69L 등 다양하며, 암젠과 미라티의 저해물질은 약 20% 정도인 G12C 돌연변이를 가진 K-ras에만 작용하며 다른 돌연변이에는 효과가 없다. 이를 극복하기 위해서는 다른 돌연변이에 특이적인 다른 돌연변이에도 모두 작용하는 약물이 필요하다.
그러나 이러한 한계도 조만간 극복될 전망이다. 미라티 세라퓨틱은 2021년 K-ras G12D 돌연변이에 특이적으로 결합하여 저해하는 화합물인 MRTX1133을 공개하였다.[14] 고해상도의 단백질-화합물 결합구조에 기반하여 디자인된 이 화합물은 시스테인과 공유결합을 하는 G12C 특이적인 화합물과 같이 공유결합으로 K-ras에 결합하지는 않지만, 단백질과의 비공유결합 상호작용을 최대한 극대화하여 G12D 돌연변이 K-ras 만을 특이적으로 저해할 수 있게 된다. 한편 다른 바이오텍 회사인 레볼루션 메디신(Revolution Medicine)은 GDP 형태로 불활성화된 상태의 K-ras에 결합하는 저해물질 대신, GTP 형태로 활성화된 K-ras와 사이크로필린 A(Cyclophillin A)라는 단백질과의 상호작용을 유도하여 활성화된 K-ras 를 억제하는 화합물을 개발하고 있다. 이들의 전략은 G12C나 G12D 등 돌연변이와 상관없이 활성화된 K-ras라면 모두 결합할 수 있으므로 보다 다양한 K-ras 돌연변이에 작동할 수 있을 것으로 기대된다.
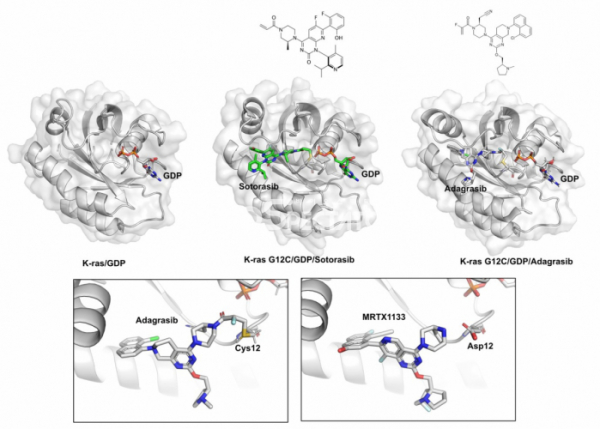
▲그림 3. (상) K-ras/GDP와 K-ras G12C 저해물질인 소토라십(Sotorasib) 혹은 아다그라십(Adagrasib)이 결합된 구조. G12C에 공유결합을 형성하여 K-ras G12C 돌연변이형을 저해하는 두 화합물은 화합물이 붙어 있지 않은 K-ras/GDP 구조에는 발견되지 않는 새로운 결합 포켓을 형성한다. (하) G12C 특이적인 저해물질인 아다그라십과 현재 개발중에 있는 G12D 돌연변이에 특이적인 저해물질인 MRTX1133. 아다그라십은 12번째 시스테인과 공유결합을 형성하는 반면, MRTX1133은 12번째 아스파르트산과 수소 결합을 형성하여 각각의 돌연변이 k-ras에 결합하여 저해한다.
이렇게 오랫동안 난공불락의 타깃으로 간주되던 K-ras 역시 오랜 시간의 노력에 의해서 이를 특이적으로 억제하는 약물이 나오게 되었으며, 이것이 가능하게 된 중요한 원동력은 역시 표적단백질의 구조에 대한 정확한 정보가 존재했다는 것이다. 특히 특정한 돌연변이를 인식하여 저해하는 화합물은 단백질의 구조 없이는 절대로 개발되지 못했을 것이다.
이번 연재에서는 이렇게 단백질 구조 정보를 기반으로 만들어진 대표적인 몇 가지의 약물의 개발 과정을 알아보았다. 다음 연재에서는 단백질 구조 기반으로 소분자 약물을 개발하는데 사용되는 여러가지 방법론에 대해서 알아보도록 하자.
참고문헌
1. Miller, M., Schneider, J., Sathyanarayana, B. K., ToTH, M. V., Marshall, G. R., Clawson, L., ... & Wlodawer, A. (1989). Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 Å resolution. Science, 246(4934), 1149-1152.
2. Roberts, N. A., Martin, J. A., Kinchington, D., Broadhurst, A. V., Craig, J. C., Duncan, I. B., ... & Machin, P. J. (1990). Rational design of peptide-based HIV proteinase inhibitors. Science, 248(4953), 358-361.
3. Krohn, A., Redshaw, S., Ritchie, J. C., Graves, B. J., & Hatada, M. H. (1991). Novel binding mode of highly potent HIV-proteinase inhibitors incorporating the (R)-hydroxyethylamine isostere. Journal of medicinal chemistry, 34(11), 3340-3342.
4. 남궁석, (2021) 바이러스, 사회를 감염하다, 바이오스펙테이터:서울 p183-207
5. Owen, D. R., Allerton, C. M., Anderson, A. S., Aschenbrenner, L., Avery, M., Berritt, S., ... & Zhu, Y. (2021). An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science, 374(6575), 1586-1593.
6. Jürg Zimmermann; Elisabeth Buchdunger; Helmut Mett; Thomas Meyer; Nicholas B. Lydon (1997). Potent and selective inhibitors of the Abl-kinase: phenylamino-pyrimidine (PAP) derivatives. , 7(2), 187–192. doi:10.1016/s0960-894x(96)00601-4
7. Schindler, T., Bornmann, W., Pellicena, P., Miller, W. T., Clarkson, B., & Kuriyan, J. (2000). Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science, 289(5486), 1938-1942.
8. Gorre, M. E., Mohammed, M., Ellwood, K., Hsu, N., Paquette, R., Rao, P. N., & Sawyers, C. L. (2001). Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science, 293(5531), 876-880.
9. O'Hare, T., Shakespeare, W. C., Zhu, X., Eide, C. A., Rivera, V. M., Wang, F., ... & Clackson, T. (2009). AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer cell, 16(5), 401-412.
10. Downward, J. (2003). Targeting RAS signalling pathways in cancer therapy. Nature reviews cancer, 3(1), 11-22.
11. Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A., & Shokat, K. M. (2013). K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature, 503(7477), 548-551.
12. Lanman, B. A., Allen, J. R., Allen, J. G., Amegadzie, A. K., Ashton, K. S., Booker, S. K., ... & Cee, V. J. (2020). Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. Journal of Medicinal Chemistry, 63(1), 52-65
13. Fell, J. B., Fischer, J. P., Baer, B. R., Blake, J. F., Bouhana, K., Briere, D. M., ... & Marx, M. A. (2020). Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer. Journal of Medicinal Chemistry, 63(13), 6679-6693.
14. Wang, X., Allen, S., Blake, J. F., Bowcut, V., Briere, D. M., Calinisan, A., ... & Marx, M. A. (2021). Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. Journal of medicinal chemistry. 65(4) 3123-3133




![[인사]한미그룹 임원 승진..김나영·최인영 부사장](https://img.etoday.co.kr/crop/268/200/2354085.jpg)


