기사본문
[BioS 레터] 'KRAS G12C 억제제'의 승인과 미래
입력 2022-09-21 10:16 수정 2022-09-21 17:12
박도현
지난해 5월18일 미국 식품의약국(FDA)는 암젠(Amgen)의 표적항암제 '루마크라스(Lumakras, Sotorasib)'를 승인한다. 승인받은 투여 대상은 KRAS G12C 돌연변이가 있는 NSCLC(non-small cell lung cancer) 환자들 중 면역항암치료를 포함하는 이전 항암치료 이력이 있는 환자들이다. 루마크라스의 승인은 표적치료제 개발 역사에 2가지 측면에서 한 획을 그었다고 할 수 있다. 첫번째로 NSCLC에서 KRAS G12C 돌연변이가 환자군에서 차지하는 비율이나 발암 및 암 진행의 중요한 부분을 차지하고 있기 때문이고, 두번째로는 타깃의 구조적 특성으로 인한 약물 설계의 어려움을 극적으로 극복해 낸 사례이기 때문이다.
루마크라스의 승인은 2003년 승인된 최초의 표적치료제인 '글리벡(Gleevec, Imatinib)' 이후로 이어져온 EGFR, ALK, MEK 등의 표적치료제에 이은 차세대 치료제로서 NSCLC를 비롯한 고형암의 생존율을 크게 높여줄 것으로 기대되고 있다. 하지만 다른 많은 표적치료제들처럼 KRAS G12C 역시 치료에 저항성이 보고되고 있으며 이를 극복하기 위한 메커니즘 스터디와 이를 근거로 한 병용치료 요법들의 시험이 이루어지고 있다. 본 글에서는 루마크라스를 중심으로 KRAS G12C 개발과 승인 그리고 KRAS G12C 억제제 저항성을 극복하기 위한 제약회사들의 전략을 바탕으로 KRAS G12C 억제제의 미래에 대해 소개하고자 한다.
1. KRAS와 암
KRAS는 GTP에 의해 활성이 조절되는 GTPase 효소로서 EGFR, MET, PDGFR 등 많은 recptor의 신호를 전달하는 하위 분자이다. KRAS는 HRAS, NRAS와 함께 RAS family를 이루며 KRAS 또한 alternative splicing에 의해 KRAS 4A와 KRAS 4B로 isotype이 나누어진다. KRAS의 isotype은 세포 내 위치와 post-translational modification이 다르게 나타나며 KRAS 4A는 저산소증과 같은 스트레스 적응에 역할을 하며 KRAS 4B는 줄기세포에서 발현율이 높다. KRAS의 돌연변이는 모든 RAS 돌연변이의 75%를 차지하고 있다. 특히 Lung adenocarcinoma 환자의 25%가 KRAS 돌연변이를 가지고 있어 KRAS의 돌연변이는 lung caner에서 높은 중요성을 가지고 있다.
KRAS mutation은 주로 12번째 G codon과 61번째 Q codon에서 나타난다. G12 codon은 C, D, A 등으로 치환되는 돌연변이가 많으며 Q61은 R, H 그리고 L 등의 아미노산으로 치환되는 경우가 많다. 폐암에서는 G12C가 전체 KRAS 돌연변이 중 가장 높은 비율인 46%를 차지하는 반면 췌장암이나 대장암에서는 G12D가 45%로 가장 많이 차지한다. 이러한 KRAS의 돌연변이는 GTPase의 기능을 약하게 하거나 GTP exchange기능을 강화함으로써 GTP에 결합하는 KRAS를 더 많이 만들고 이로 인해 KRAS의 활성이 높아진다. 높은 활성의 KRAS는 ERK로 대표되는 하위 분자의 활성을 강화시켜 세포의 생존, 분열을 촉진시키고 면역세포 회피능력을 강화한다.
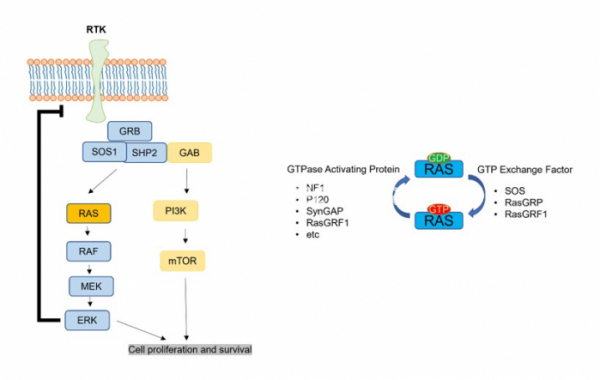
▲그림 1. RAS 신호경로와 RAS 활성 사이클
2. KRAS G12C 억제제의 개발
2-1 Kevan Shokat과 ARS-3620
KRAS 돌연변이의 중요성에도 불구하고 KRAS 억제제의 개발은 수십년간 이루어지지 못했다. 첫번째 이유는 KRAS와 GTP 간 결합의 Kd 값이 pM 범위로 아주 강하기 때문에 GTP와 결합력을 경쟁할 수 있는 저분자 화합물을 발굴하기 어려웠기 때문이고 두번째로는 KRAS가 구조상 allosteric inhibitor가 결합할 만한 포켓이 없는 평이한 구조를 가졌기 때문이다. 이러한 어려움 때문에 저분자 화합물 억제제 개발은 쉽게 이루어지지 않았다.
이러한 문제는 2013년 UCSF의 Kevan Shokat 교수가 출판한 논문을 통해 실마리가 풀려나가기 시작했다. 이 연구에서 연구팀은 G12C mutation이 있는 KRAS의 구조를 밝혔는데 KRAS의 beta-sheet의 아랫부분의 저분자 화합물이 결합할만한 allosteric pocket을 발견했다. 이 allosteric pocket은 KRAS가 GDP 상태일 때만 노출되는 특징을 가졌는데 pocket에 위치한 cystein에 공유 결합을 할 수 있는 저분자 화합물이 불활성화 KRAS인 KRAS-GDP를 유지하게 함으로써 KRAS G12C의 활성을 억제하는 메커니즘을 처음으로 제시하였다. 이 발견을 바탕으로 Shokat교수는 아락세스 파마(Araxes pharma)를 창업하였고 자회사인 Wellspiring Bioscience를 통해 그의 연구에 기반한 KRAS G12C 억제제 개발을 지속하였다.
바로 1년 후 KRAS G12C 억제제의 가능성을 본 J&J는 아락세스와 파트너십을 맺고 임상개발을 추진하였다. 하지만 현재 J&J의 KRAS G12C 억제제(ARA-3620/JNJ-74699157)는 phase I이 끝난 현재 안전성 문제로 개발이 중단된 상태다.
KRAS 억제제를 개발할 수 있는 핵심적인 발견이 이루어졌지만 KRAS의 돌연변이는 KRAS의 GTP결합을 높여주기 때문에 KRAS-GDP에만 결합할 수 있는 억제 메커니즘에는 여러가지 회의적인 시각이 많았다. 다행스럽게도 G12C 변이는 다른 변이에 비해 여전히 GTP-GDP의 사이클을 상당히 유지하기 때문에 GDP형태의 KRAS에 결합하는 억제제가 잘 작동할 수 있었다. 그럼에도 불구하고 여전히 KRAS G12C는 GTP형태가 많고 G12C 외에 다른 변이에서는 KRAS-GTP 비율이 훨씬 높기 때문에 KRAS가 활성화되어있는 GTP 형태에서도 결합하여 억제할 수 있는 억제제 개발이 필요성이 대두되고있다. 그리고 이에 대한 해답은 뒤에 언급될 레볼루션 메디슨(Revolution medicine)사의 KRAS-on 억제제가 줄 수 있다는 기대감이 만들어지고 있다.
2-2 암젠의 Sotorasib
비슷한 시기에 암젠도 KRAS G12C 억제제 개발에 뛰어들었다. 암젠은 Carmot Therpaeutics와 2014년에 KRAS G12C 억제제에 대한 공동연구를 시작하였는데 Carmot의 lead compound 발견 기술이 사용되었고 여기서 나온 물질들을 암젠에서 리드 화합물로 선정하는 과정을 2017년 12월까지 이어나갔다. 2019년 ESMO에서 암젠은 자사의 개발 물질인 AMG510가 임상 I/II단계에서 13명의 환자의 54%가 PR을 보인 결과를 발표한다. 연구개발 시작 약 4년만에 임상1상 결과를 내놓는 아주 빠른 속도를 보였는데 이로 인해 미라티(Mirati)와 J&J를 비롯한 다른 경쟁사들에 비해 더 빠른 승인을 받아냈다. AMG510은 성분명 Sotorasib으로 명명되었고 이후 루마크라스(Lumakras)라는 판매 이름을 갖게 되었다.
이 승인은 CodeBreaK100이라고 명명된 2상의 임상시험의 결과를 바탕으로 이루어졌는데 이 시험에서 KRAS G12C mutation을 가지고 있으며 이전에 면역/화학 치료를 받은 이력이 있는 환자들이 하루 960mg의 약을 복용하였다. 전체 124명의 환자 중 ORR(Overal Response Rate)이 36%였고 DCR(Disease Control Rate)은 81% 나타냈다. DoR(Duration of Response)의 중간값은 10개월이었다. 이러한 효능과 함께 보고된 부작용은 설사, 복통, 피로 등이 있으며 약 9%의 환자가 이러한 부작용으로 인해 치료를 받지 못했다. 현재 Sotorasib은 NSCLC 환자에게서 docetaxel과 효능을 비교하는 3상시험 CodeBreaK200을 진행하고있다. 이외에도 KRAS G12C의 억제가 환자의 종양 면역환경을 종양 공격성향으로 바꾼다는 pre-clnical과 clinical 결과를 바탕으로 면역항암제인 anti PD-1 항체와의 병용요법 시험도 실시되고 있다.
2-3 미라티(Mirati)의 Adagrasib
미라티의 Adagrasib은 암젠의 Sotorasib과 실질적으로 승인 1호 경쟁을 벌였던 약물이다. Adagrasib은 비임상 스터디에서 AMG510과 비슷한 정도로 KRASG12C를 억제하였고 체내 반감기는 24시간 가까이돼 AMG510 대비 장점이 되는 약물성도 지녔다. 하지만 개발 속도면에서 Adagrasib은 뒤쳐지기 시작했고 최근 발표된 임상 2상 결과에서 40.6%의 ORR(Sotorasib은 43%)을 보였음에도 후발 주자라는 측면에서 더 나은 유효성을 보이지 못한 한계를 보였다. 약효 지속시간인 DoR은 8.5개월로 Sotorasib의 12.5개월에 비해 적게 나타나 미라티의 주가가 당일 30% 이상 떨어지기도 하였다. 현재 미라티는 Adagrasib이 높은 혈뇌장벽(Blood Brain Barrier) 투과도를 가지고 있다는 점을 강점으로 내세우며 Sotorasib과 차별화를 꾀하고 있다.
실제 보고된 바에 따르면 Adagrasib은 마우스 뇌전이 모델에서 cellular IC50 이상을 상회하는 CSF(Cerebrospinal Fluid)농도를 보였고 실제로도 마우스 모델의 전이된 종양에서 바이오 마커인 pERK가 adagrasib에 의해 감소함을 보였다. 임상2상에서도 CNS로 전이가 되고 치료이력이 없는 25명의 환자들이 adagrasib을 600mg BID로 투여받을 경우 adagrasib의 CSF/free plasma ratio가 0.47 정도로 높게 측정되었고 IC disease control rate도 84%로 측정되었다. 이러한 특징은 Adagrasib에게 큰 장점으로 작용하는데 KRAS mutation이 있는 NSCLC 환자의 경우 높은 비율(27-42%)로 CNS전이가 일어나기 때문이다. 뇌 투과성을 높이면서 약효를 유지하는 약물 개발은 더욱이 어려운 만큼 Mirati는 이러한 점을 차별화로 내세우는 전략을 지속할 가능성이 높다.
2-4 그 외의 KRAS G12Ci
제넨텍(Genentech)의 GDC-6036, 자코바이오(Jacobio Pharmaceuticals)의 JAB-21822, 일라이릴리(Eli Lilly)의 LY3537982, 베링거인겔하임(Boehringer Ingelheim)의 BI-1823911 등의 물질들이 현재 임상1/2상에 들어가 있다. 이들 물질들은 단독요법과 함께 CDK4/6i, anti PD-1, SOS1i, SHP2i 혹은 EGFRi와 병용요법들의 임상시험이 진행중에 있다.
3. KRAS G12C 억제제의 저항성
KRAS G12C의 반응률은 다른 표적항암제인 Osimertinib이나 imatinib(각각 EGFR, BCR-ABL 타깃)에 비해 높지 않은데 그러한 이유의 일부는 약물의 저항성 때문인 것으로 보인다. 세포 모델에 Sotorasib이나 Adagrasib을 처리하면 빠른 시간 안에 RAS 경로를 억제하지만 시간에 지남에 따라(6시간 이상) RAS 경로가 다시 활성화되고 바이오마커인 pERK의 레벨이 약물처리 전에 근접한 수준으로 회복됨을 볼 수 있다. 이러한 저항성의 일부는 negative feedback에 의한 RTK(receptor tyrosine kinase)의 재활성화와 WT KRAS의 역할 증가에 의한 것으로 알려졌다. 임상에서 나타나는 약물의 저항성이나 재발이 발견된 환자에서 ctDNA(circulating tumor DNA)를 이용해 sequencing을 했을 때 FGFR이나 EGFR과 같은 RTK의 돌연변이가 새로 발견되거나 증가했으며 특히 Y96D와 같은 KRAS on-target mutation도 여럿 발생하는 것으로 알려졌다.
이러한 on-target mutation은 약물이 결합하는 단백질의 부위를 변형시켜 약물의 결합력을 떨어뜨리거나 KRAS가 상위 분자와 무관하게 항상 GTP상태에 있게 만듦으로써 약물에 저항성을 갖게 한다. 이에 따라 KRAS G12C 억제제의 한계를 극복하려는 노력들이 이미 시도되고 있고 이들 중 일부는 이미 임상에 진입했다. Negative feedback에 의한 RTK pathway의 활성화 증가는 SHP2, SOS1 혹은 PI3K와 같은 단백질들을 타깃으로 하는 표적 항암제들과의 콤비에 대한 필요성을 불러일으켰다. 그리고 on-target mutation 역시 KRAS upstream 저해제를 통한 극복을 생각해 볼 수 있으나 G12R이나 Q61X와 같은 상위 분자와 무관하게 항상 KRAS를 활성화시키는 돌연변이와 기존 억제제의 결합을 방해하는 돌연변이도 발생된다는 점에서 다양한 mutation이 있는 KRAS도 억제할 수 있는 pan-KRAS 억제제 대한 개발도 시도되고 있다.
4. SHP2
RAS pathway는 암 생물학에서 아주 중요한 부분을 차지하고 있기 때문에 이를 구성하는 분자들의 대부분을 타깃으로 하는 약물의 개발이 집중적으로 이어져 왔으며 MEKi, BRAFi와 같은 표적항암제들은 이미 환자에게 적용되었다. 이에 반해 SHP2와 SOS1 혹은 GRB2 같은 상위 분자들은 MEK, RAF나 ERK와 같은 하위 분자들에 비해 덜 중요하게 여겨졌지만 최근에는 기존 RAS pathway 억제제들의 저항성에 기인하는 주요 요인으로 주목을 받고 있어 개발이 활발히 진행되고 있다.
SHP2는 phosphatase로서 EGF 등과 같은 liand에 의해 활성화된 RTK에 결합하여 스스로가 활성화된 후 하위 분자들의 활성을 조절한다. SHP2 자체의 돌연변이와 과활성이 여러 암종에서 발견되었고 또한 RAS pathway 억제제에 의한 저항성 상황에서 phosphorylation과 이에 따른 활성 증가가 보고되었으며 SHP2의 발현을 줄이거나 활성을 억제할 경우 저항성을 극복할 수 있다는 보고가 이루어졌다. 하지만 phosphatase의 경우 catalytic site를 타깃 했을 때 다른 phosphatase와 구별되는 selectivity를 갖기가 어려워 억제제의 개발이 활발히 이루어지지 않았다.
이런 가운데 노바티스(Novartis)에서는 SHP2의 inactive conformation을 고정시키는 molecular glue를 개발함으로써 SHP2 억제제를 발굴해 내었는데 현재 임상물질인 TNO-155가 mono therapy, 그리고 KRAS G12Ci와 anti-PD1과 같은 다른 항암제와의 병용 요법에 대한 임상1상이 진행 중에 있다. 이후 SHP2에 대한 관심은 계속 증가하여 현재 Revolution medicine(RMC-4630), Genentech/Relay therapeutics(GDC-1971) 등이 SHP2억제제를 개발하여 임상단계에 들어갔으며 후속 주자들의 개발도 활발히 이루어지고 있다.
5. SOS1
SOS1은 GEF(GTPase Exchange Factor)로서 KRAS를 비롯한 RAS의 GTP loading을 촉진하는 역할을 한다. SOS1 역시 SHP2와 마찬가지로 여러 암에서 활성 증가가 관찰되었고 높은 비율은 아니지만 여러 암들, 특히 혈액암에서 돌연변이가 관찰되었다. 바이엘(Bayer)은 2019년 논문을 통해 SOS1에 결합하여 SOS1과 KRAS와의 결합을 방해하는 MoA를 가진 BAY-293을 발표하였다. 이후 Bayer는 이 SOS1억제제의 개발을 진행하지 않은 것으로 보이나 다른 회사들의 SOS1 억제제 개발의 시발점이 되었다.
베링거인겔하임(Boehringer Ingelheim)은 2020년 BAY-293과 같은 MoA의 BI-3406을 논문을 통해 발표하였으며 현재 BI-1701963은 KRAS mutation이 있는 환자군에서 단독과 베링거인겔하임의 KRAS G2Ci인 BI-1823911 혹은 Adagrasib과의 병용투여를 포함하는 임상1상이 진행 중에 있다. 이외에도 미라티 역시 Adagasib과의 병용을 염두에 둔 MRTX-0902의 임상을 시작하였으며 최근에 나온 발표를 토대로 볼 때 두 물질의 병용을 KRAS mutation을 가진 NSCLC 환자 중 뇌전이가 일어난 환자들로 타깃할 가능성이 높다. 이 외에도 Revolution medicine(RMC-5845)과 ERASCA(unknown)등이 SOS1 개발을 진행하고 있다. SOS1은 특정 mutation이 있는 KRAS 뿐 아니라 모든 타입의 KRAS와 SOS1과의 결합을 저해하기 때문에 베링거인겔하임은 SOS1 억제제를 Pan-KRAS inhibitor라고 명명하고 있다. SOS1을 억제할 경우 KRAS의 GDP상태를 늘려주기 때문에 KRAS-GDP상태에만 결합하는 Sotorasib과 Adagasib의 효능을 높일 것으로 기대하고 있다. 또한 SOS1은 KRAS의 상위에 위치하기 때문에 KRAS G12Ci에 의한 on-target mutation의 활성도 억제할 수 있는 것으로 보고되었다.
6. RAS on inhibitor
지금까지 개발되거나 시도되는 KRAS inhibitor는 G12C mutation이 생겼을 때 나타나는 결합 부위와 거기에 위치한 cystein 잔기를 통한 공유 결합을 이용하는 방식을 취했다. 그렇기 때문에 낮은 비율의 GDP상태의 KRAS에 결합만 할 수 있었고 MoA측면에서 한계를 가질 수밖에 없었다.
하지만 레볼루션 메디슨은 GTP를 결합한 on 상태의 KRAS에도 결합하여 활성을 억제할 수 있는 억제제를 개발했다. RMC-6291은 KRAS G12C와 CyclophilinA에 결합해 Tri-complex를 만들고 이러한 구조는 KRAS가 하위 분자들과 결합하는 것을 방해하기 때문에 KRAS 활성이 억제된다. 이와 같은 방식은 상당히 독창적이면서 동시에 역사에 기인한다. 레볼루션 메디슨이 밝히듯이 mTOR와 FKBP12 사이에 결합하여 tri-complex를 만드는 mTOR의 natural inhibitor인 Rapamycin에서 메커니즘을 차용했기 때문이다. RMC-6291은 여전히 KRAS G12C 돌연변이에 선택적이면서 기존의 GDP 상태의 KRAS G12C에 결합하는 억제제보다 월등한 효과를 전임상 모델들에서 보여줬다.
이뿐 아니라 같은 방식을 이용해 KRAS G12D(RMC-9805), KRAS G12C(RMC-8839)그리고 Pan-KRAS mutation(RMC-6236)까지 다양한 타입을 공략할 수 있는 억제제들을 개발하고 있다. 2022년 6월 28일 레볼루션 메디슨은 RMC-6246의 첫번째 환자 투여가 이루어졌다고 발표를 하였다. MoA 측면에서 장점을 가진 KRAS on 억제제가 기존의 KRAS off 억제제에 비해 전임상에서 보여준 것만큼 임상에서 효능을 보여줄지가 특히 기대되는 부분이다. 특히 독성 이슈에 대해서도 이점이 있다면 KRAS on 억제제들은 후발주자임에도 차세대 KRAS 억제제로서 시장을 선점할 가능성이 높을 것이다.
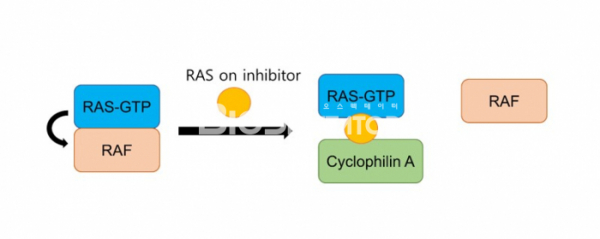
▲그림2. RASon 억제제의 메커니즘
KRAS G12C 억제제의 승인은 수십년간 타깃이 되지 못했던 암 생물학의 핵심 분자인 KRAS를 드디어 공략할 수 있다는 큰 기대감을 주었다. 하지만 동시에 환경에 적응하고 살아남는 암세포의 특성은 KRAS G12C 억제제에도 영향을 미쳐 Sotrasib과 Adagrasib의 약효에 한계를 가져왔다. RAS pathway에 속한 다른 분자들을 타깃하는 방법 그리고 RASon 억제제와 RAS-degrader 등의 다양한 MoA를 가진 약물들이 암세포의 저항력이 만들어낸 약효의 한계를 극복할 수 있을지 아니면 또 다른 새로운 전략이 필요할지에 대한 방향이 앞으로 중요할 것이다.
참고문헌
1. Mirati press release
2. Amgen press release
3. Revolution medicine press release
4. Moore et. al. 2020 Nat. Rev. Drug. Discov. 19(8) 533-552
5. Blair et al. 2021 Drugs 81, 1573-1579
6. Sabari et al. 2022 Cli. Can. Res.
7. Koga et al. 2021 J. Thor. Onc. 16(8) 1321-1332
8. Awad et al. 2021 NEJM. 384 2382-2393
9. Mullard et al. 2019, Nat. Rev. Drug. Discov. 18, 887-891
10. Fuenta et al. 2022, Front. Onco., 11, 792635
11. De´sage et al. 202, Front. Onco., 12, 796832
12. Janes et al. 2018 Cell. 172 978-89
13. LaMarche et al. 2020, J. Med. Chem. 63, 22, 13578–13594
14. Hofmann et al. 2021 Cancer Discov. 11 142-57
15. Shen et al. 2020 Eur J. Med. Chem. 109 11217
16. Kessler et al. 2020 Curr. Opi. Chem. Biol. 62 109-118
17. Wang et al. 2022 The Oncologist 27(7) 536-553







