기사본문
A different view on the approval of aducanumab
입력 2021-07-08 13:33 수정 2021-07-08 13:34
by Sungmin Kim
The U.S. Food and Drug Administration's (FDA) marketing approval for the first Alzheimer's disease drug 'Aduhelm (Aducanumab)' was 'a radical decision beyond innovation'. Immediately, three of the 11 advisory members of the Peripheral and Central Nervous System Drugs Advisory Committee (PCNS) resigned one after another. FDA has come to stand alone in the face of the responsibility of fully accepting changes and repercussions that go beyond the scope of innovation.
The FDA decision is shockingly acceptable in two respects. First, the accelerated approval was granted based on a surrogate called amyloid plaque in the brain. Even though there was a prospect that the FDA would grant conditional approval of Aduhelm, The biomarker-based marketing approval, not the cognitive improvement, which is the primary endpoint set in phase 3 clinical trial, is an unexpected result. Second, it has a broad indication on the product label. A rather vague license was granted as “a prescription drug to treat Alzheimer’s disease patients,” rather than for the early stage Alzheimer’s disease, which is mild cognitive impairment (MCI) or mild stage, the subject of which Biogen has conducted phase 3 clinical trials. Per Biogen, the prescription depends on the judgment of the doctor. Rather, this was also contrary to the industry's prediction that it could be granted a marketing authorization conservatively by reducing the number of prescriptions.
Controversy aside, the dice have already been cast. The FDA made this marketing authorization decision based on the success of an independent phase 3 clinical trial, whether this decision is right or wrong, and real-world data will now prove the decision. Why did the FDA, a regulatory authority that has no choice but to make a conservative decision, take such a risk? Despite overwhelming opposition, why did the FDA not take a step back and make the most controversial decision to 'Approve the marketing of Aduhelm' in history? In order to understand and move forward one step further, it is necessary to look into the complicated process of contemplation that the FDA has put forward in the field of new drug development for Alzheimer's disease.
“Three-year gap”...Indication label for even early patients in mind?
On February 15, 2018, the FDA published a draft amendment for the first time in five years to aid the clinical development of Alzheimer's disease and other neurological diseases for which there is no cure. The key point was to select patients based on biomarkers in the initial clinical trials for Alzheimer's disease and to grant accelerated approval as the primary endpoint is achieved. At the time, when the FDA announced this in the industry, it was welcoming, expecting that the development of Alzheimer's disease treatment and early diagnosis would be activated.
As the scientific understanding of AD has evolved, efforts have been made to incorporate in clinical trials, to varying degrees, the use of biomarkers reflecting underlying AD pathophysiological changes and the enrollment of patients with AD at earlier stages of the disease, stages in which there may be no functional impairment or even no detectable clinical abnormality
Regarding the background of the announcement of the draft, the FDA said, “As the scientific understanding of AD has evolved, efforts have been made to incorporate in clinical trials, to varying degrees, the use of biomarkers reflecting underlying AD pathophysiological changes and the enrollment of patients with AD at earlier stages of the disease, stages in which there may be no functional impairment or even no detectable clinical abnormality. These efforts are particularly important because of the opportunity to intervene very early in the disease process that AD provides... (omit)... It is obvious that delaying, or, preferably, halting or reversing, the pathophysiological process that will lead to the initial clinical deficits of AD is the ultimate goal of presymptomatic intervention, and treatment directed at this goal must begin before there are overt clinical symptoms."
Around this time, the movement to define Alzheimer's disease based on biomarkers began, supporting the FDA's policy. Shortly after the FDA released the draft, in April, the National Institute of Aging and the Alzheimer's Association (NIA-AA) under the National Institutes of Health (NIH) announced that they would define Alzheimer's disease based on three biomarkers- amyloid, tau, and neurodegeneration (A/T/N)- rather than symptoms.
However, no progress has been made so far after the FDA published these draft amendments. Since then, there has been no company that the FDA has announced an amendment to the content or that they will conduct clinical development using the biomarker as the primary endpoint. So far, there has been a gap of over 3 years after draft amendments.
The reason for this is that the FDA at the time indicated the possibility of recognizing biomarkers as clinical satisfaction endpoints was a very early Alzheimer's disease patient, before the MCI (or prodromal), who showed pathological changes but had no clinical symptoms. In order to proceed with these patients, preventive trials must be carried out, and a long time and huge amount of money must be invested. In addition, even if early diagnosis products for Alzheimer's disease are released, it is difficult to be widely distributed in the absence of a cure.
If you can guess the approximate numbers, the phase 1 clinical trial of Aducanumab started in 2011, and based on the positive phase 1b clinical results, without a phase 2 clinical trial, since 2015, the phase 3 clinical trial was conducted with about 3,300 patients in 20 countries around the world and ended in 2019. The total cost of clinical administration in this process is estimated to be $1.63 billion (from the Evaluate Pharma data). In fact, with Big Pharma neglecting to develop a new drug for Alzheimer's disease due to a series of failures, it seems difficult to decide to risk preventive clinical trials that require a four to five-year drug administration period.
Maybe that's why. The FDA did not narrow Aduhelm's license label to patients with mild cognitive impairment and mild staged patients, but granted a marketing license to prescribe early patients without cognitive impairment. According to Biogen, more than 900 sites in the U.S. are now ready to prescribe Aduhelm.
'Best-in-class' for now?
Data and development status of competing drugs are also believed to have played an important role in the FDA's decision to approve Aduhelm. In this regard, it is necessary to keep an eye on the development of amyloid and tau antibody candidate materials that conform to biomarker treatment strategies.
First of all, according to the biomarker data that the FDA approved to market Aduhelm, amyloid plaque in PET images decreased by 25 to 30% when 10mg/kg of Aducanumab was administered for 78 weeks in Phase III clinical trials.
Next are Ezai-Biogen's "Lecanemab" (BAN2401) and Lilly's "Donanemab" (Donanemab) in the late clinical development phase, which is close to commercialization. Lecanemab is an antibody that binds to the toxic amyloid oligomer–flake stage similar to Aducanumab, and Donanemab is an N3pG target antibody that binds selectively only to the amyloid plaque, the toxic amyloid's "reservoir".
Comparing the biomarker results, in clinical trials, when Lecanemab 10mg was administered weekly for 78 weeks to early-stage Alzheimer's disease patients, amyloid plaques decreased by 22% compared to pre-administration, and when 10mg was administered every other week, the amyloid plaques decreased by 30%. Similar results to Aducanumab. Lecanemab is currently in phase 3 clinical trials, and the clinical trials will end in 2024.
The data worth paying attention to is Lilly's Donanemab. In phase 2 of TRAILBLAZER-ALZ clinical trial, Donanemab showed a rapid drug response in which amyloid plaques decreased by more than 37% from the 24th week of dosing, and about 46% decreased at the 76th week(SUVR parameter). Lilly amended the protocol for the second TRAILBRLAZER-ALZ2 clinical trial for Donanemab, expanded the number of patients from 500 to 1500, which is currently undergoing; the first efficacy results are expected to be announced in 2023. However, recently reversing the original plan, Lilly plans to seek accelerated Food and Drug Administration approval and intends to submit a biologics license application (BLA) for donanemab under the accelerated approval pathway later this year.
However, both drugs have announced positive phase 2 clinical results so far, but on the other hand, doubts in the industry remain as they have not significantly slowed cognitive impairment or obtained consistent data from various indicators.
Lecanemab announced that the Data Monitoring Committee (DMC) recommended clinical suspension because there was no possibility of significant differences compared to placebo in the 12th month, but Ezai confirmed the possibility and confirmed statistically significant differences in the 18th month. Next, Lilly announced in January this year that Donanemab delayed cognitive impairment by 32% compared to placebo after 76 weeks of medication in Phase II clinical trial. However, it has been controversial that the difference between the two groups in clinical data disclosure has not been significant, and that there has been no difference in secondary endpoints. In other words, both Lecanemab and Donanemab have seen positive signals, but it is difficult to give a definite answer to the clinical benefits.
In addition, Roche is undergoing phase 2 clinical trial with "RG6102" (brain shutter gantenerumab), which has a TfR1 shuttle that improves blood barrier (BBB) permeability. According to Roche's recent data released at the ADPD conference, RG6102 increased the cerebrospinal fluid/plasma ratio (CSF/plasma ratio) to gantenerumab without BBB shuttle by about eight times in clinical 1 phase in healthy subjects.
Drug development targeting tau, another key pathological protein in Alzheimer's disease, is still in the early phase. Big Pharma, such as AbbVie, Biogen, and Roche, as leaders, failed all phase 2 clinical trials of Tau antibody targeting degenerative brain diseases including Alzheimer's disease, and there was no data showing positive signals.
In conclusion, unless any changes are sought, it is difficult to make a breakthrough in the field of Alzheimer's disease within a few years. In addition, the fact that even leading amyloid drugs have uncertainty due to mixed data seems to have supported the decision to approve it.
A precedent for surrogate marker-based acceleration approval
This time, when the FDA made a decision to approve Aduhelm based on the surrogate marker, attention was focused on the case in which the FDA previously granted a marketing approval based on the surrogate marker. In which cases was the surrogate marker clinical endpoint applied, and what was the result?
The FDA-defined surrogate endpoint is a marker measured in a laboratory, such as radiographic images, physical indicators, and so on, which does not in itself address clinical benefits. The guidelines state that there are two cases of surrogate markers. Surrogate markers: ▲ clinical benefits can be predicted, and if used to support existing drug and biopharmaceutical approvals ▲ have a reasonable chance of predicting clinical benefits and can be used to support drug and biopharmaceutical acceleration approvals. Examples of the former are indicators of blood sugar, blood pressure, and LDL used in cardiovascular diseases, and Aducanumab is the latter case.
Then let's take a closer look at the latter case. We are looking at three cases: cancer disease, HIV, and Duchenne muscular dystrophy (DMD).
In fact, the field in which surrogate markers are most actively applied is in anticancer drugs. In anticancer drugs, the “gold standard” is to confirm improvement in the patient's OS rate after 5 years, but it is difficult to apply it to actual clinical development. In response, the FDA introduced an accelerated approval system using surrogate markers in 1992, and since then, indicators that can predict the prognosis of patients according to cancer types such as overall response rate (ORR), progression-free survival (PFS), and disease-free survival (DFS) have been used.
In fact, the FDA approved approximately 194 drugs based on surrogate marker clinical endpoints over 28 years, 89 were accelerated, and were the first cases to be applied by surrogate markers in certain cancers (1992.01–2019.07) (doi: 10.1001/jamainternmed.2020.1097). Although surrogate markers advance clinical development of new drugs and promote more drugs to be marketed, controversy is still ongoing over the link between surrogate markers and OS indicators in clinical trials. For a close example, in April, the FDA held an oncologic drugs advisory committee (ODAC) for three days to decide on final approval for PD-(L)1 immunocancer drugs that received acceleration approval, and recommended maintenance of four out of six cases and withdrawal of two cases.
Surrogate markers are indicators that can change as scientific understanding develops. According to FDA reports, surrogate markers have advanced in HIV drug approval, using CD24/p24 antigens since 1991 and placing importance on the CD4/HIV-RNA and CD4 indicators in 1996. In this process, various drugs such as diddanosine (ddI), zalcitabine (ddC), and ritonavir (RTV) were released, and drugs that did not show clear efficacy disappeared.
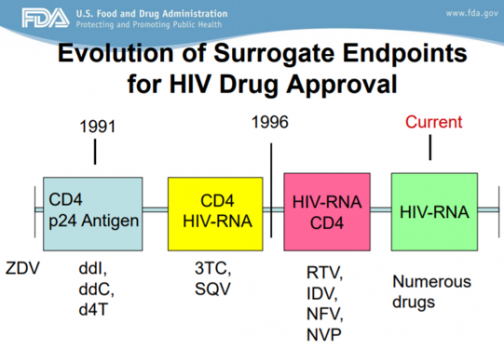
Applying this, Aduhelm's approval can be seen as an opportunity to start researching pathological biomarkers in the field of developing new drugs for Alzheimer's disease.
In neurological diseases, Sarepta's DMD treatment Exondys 51 was also a drug that caused a lot of controversy during the marketing approval process. In 2016, Exondiy51 received a negative recommendation from the FDA advisory committee, and even within the FDA, there were sharp disagreements within the FDA, leading to the resignation of the executive director. After the pros and cons debate, the final marketing approval decision was made on the condition of proving the efficacy of the drug in confirmatory clinical trials, and the voices of patient groups at the time also played a decisive role in drug marketing. Considering that there is no treatment for Duchenne muscular dystrophy, the FDA approved conditional approval based on biomarker data that improved dystrophin levels at 180 weeks after administration to 0.22 to 0.303% compared to normal subjects. Whether the actual dystrophin biomarker leads to clinical benefit has yet to be concluded.
Sometimes the evaluation of controversial drugs is decided in the marketplace. It also accelerates competition for development of latecomers. Sales of Exondys 51 in 2019 were $381 million. Sarepta then received marketing approvals for two DMD products, "Exondys 53" and "Amondys 45". Last year, "Viltepso"(Viltoarsen) by NS Pharma was released as a competitive drug for Exondis 53.
"The next nine years."
What process remains to be done? As the FDA makes a conditional decision on Aduhelm's approval, Biogen must proceed with Phase IV of the corroborative clinical trial to demonstrate drug efficacy in patients with Alzheimer's disease. The FDA said it could withdraw the drug from the market if it fails to meet the expected results of confirmation.
According to the industry, Biogen has to prepare a clinical plan by August, complete the clinical trial by 2029, and submit a final report to regulators in 2030. The next nine years are also an opportunity for Biogen to generate tens of billions of dollars in sales, but on the other hand, it has a big challenge to solve.
Currently, three FDA-approved amyloid PET tracers, which cost $4,000 to $5,000, while they are not covered by insurance. In response, Biogen also presented the results of analyzing the correlation between cerebrospinal fluid (CSF Aβ42, Aβ40, p-tau181, t-tau) and amyloid PET results at the ADPD conference held in April to improve patient access. According to Biogen, the CSF Aβ42/Aβ40 ratio showed the highest accuracy among various indicators.
The biggest obstacle in the immediate commercialization process is the administration cost of $56 million per year for Aduhelm. The price was well above the $2,500-8300 suggested by the Center for Drug Evaluation and Research (ICER), a non-profit organization in the United States. Despite these estimates, ICER estimated annual sales of Aducanumab at $50 billion. Currently, its benefits vs. side effects is not clear, so the question remains as to how much the insurance company can pay. In the mid- to long-term, Biogen must also address the issue of how to find and diagnose more early-stage Alzheimer's disease patients, and the insurance coverage of PET/CSF-based diagnostic methods to increase access to these patients.
Production capacity?…“Third CMO possibility” was mentioned
There are 6 million people with Alzheimer's disease in the United States. Among them, Biogen estimates the number of MCI or mild amyloid-positive patients to be between 1 and 2 million. In addition, it is currently under review for marketing approval for Aduhelm in countries such as Canada, Europe, Switzerland, Japan, Australia and Brazil.
Biogen explained there are 2 production sites with a total production capacity of 263,000 liters. First, it is producing Aducanumab DS and DP at the Research Triangle Park (RTP) facility in North Carolina, USA, and Biogen invested $2 billion in the solothurn facility in Switzerland to prepare for an increase in Aducanumab production scale and efficiency. The facility was designed for high-efficiency production of Aducanumab, with a production yield of 10 g/L. At the end of this year, it plans to submit an application for permission to produce Aducanumab to the FDA.
Biogen said it could produce Aducanumab, which will serve about 1 million patients at the Solothurn site, depending on additional demand in the future. Furthermore, Biogen mentioned that it is considering a third-party CMO for additional volume demand. This is positive news for Samsung Biologics, a partner of Biogen and the world's top-tier production capacity. In addition, Biogen also talked about continuously improving production efficiency and increasing existing site facilities.
Finally, it is necessary to reflect on the words of Patrizia Cavazzoni, director of the Center for Drug Evaluation and Research (CDER), in the official document for approval of the marketing authorization for Aduhelm.
"FDA will continue to monitor Aduhelm as it reaches the market and ultimately the patient’s bedside. Additionally, FDA is requiring Biogen to conduct a post-approval clinical trial to verify the drug’s clinical benefit. If the drug does not work as intended, we can take steps to remove it from the market. But hopefully, we will see further evidence of benefit in the clinical trial and as greater numbers of people receive Aduhelm. As an agency, we will also continue to work to foster drug development for this catastrophic disease."







