기사본문
HLB, '리보세라닙' 제조소 FDA 실사종료서한 "수령"
입력 2026-07-15 09:36 수정 2026-07-15 10:08
바이오스펙테이터 이효빈 기자
cGMP 실사결과 'VAI(자발적 개선권고조치)'로 최종 분류.."FDA 입장 확인 위한 공식질의 추진"

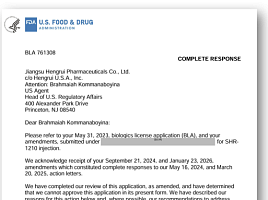
HLB의 미국 자회사 엘레바테라퓨틱스(Elevar Therapeutics)는 지난 14일 파트너사인 항서제약(Jiangsu Hengrui Pharmaceuticals)으로부터 미국 식품의약국(FDA)의 ‘리보세라닙(rivoceranib)’ 원료의약품 제조소에 대한 일반 cGMP 실사종료서한(close-out letter)을 수령했다고 15일 밝혔다.
회사에 따르면 미국 식품의약국(FDA)은 해당 제조시설의 cGMP 실사 결과를 'VAI(자발적 개선권고조치)'로 최종 분류했으며, 수령받은 서한에는 "VAI 분류 자체가 해당 제조시설과 연계된 현재 진행중인 허가 신청에 대한 FDA의 평가에 영향을 미치지 않는다"고 명시돼 있다.
FDA는 앞서 CRL에서 리보세라닙과 ‘캄렐리주맙(camrelizumab)’ 병용요법의 간암 1차치료제 승인거절 사유가 진차오 사이트의 일반 cGMP 실사 결과에 따른 것이라고 알린 바 있다. 엘레바는 해당 실사가 VAI로 종결된 데 따른 FDA의 입장을 확인하기 위해 타입A 미팅과 함께 추가적인 공식 질의를 추진할 계획이다.
완제의약품(DP) 제조시설에 대한 Form 483 역시 원료의약품 제조시설과 유사한 범주의 지적사항이 포함된 것으로 알려져 있으며, 항서제약은 오는 24일까지 답변서와 시정·예방조치(CAPA) 계획을 FDA에 제출할 예정이라고 회사는 설명했다.