기사본문
[남궁석의 신약연구史]AIDS 치료제 개발, 칵테일요법
입력 2019-08-14 10:28 수정 2019-08-14 10:28
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
1990년말에 이르러 전세계적으로 공식적인 AIDS 환자의 추계는 30만7000명, 그리고 실제 환자는 약 100만명으로 이를 것으로 추산되었고, 세계적으로 HIV 감염자의 수는 800만~1000만명에 이를 것으로 추산되었다. AIDS에 대한 공포는 세계적으로 확산되었다. 그러나 1990년대 중반에 등장된 항바이러스 요법, 특히 고활성 항레트로바이러스 치료법(highly active antiretroviral treatment. HAART)이라고 불리는 항바이러스 요법의 등장은 HIV 감염을 통제와 관리가 가능하도록 만들었다. 이번 연재에서는 HAART을 구성하는 여러 가지 화학저해제들의 개발 과정을 알아보도록 한다.
HAART의 구성 요소
HAART로 불리는 항레트로바이러스 바이러스 치료법의 특징은 레트로바이러스의 생육 사이클의 여러 단계를 저해하는 여러개의 약물을 동시에 투여하여 바이러스의 복제를 억제한다. 이들을 구성하는 약물은 크게 다음의 종류로 나뉜다.
- 역전사효소 저해제(Reverse Transcriptase Inhibitors): HIV가 속해 있는 레트로바이러스의 생육 사이클에서 가장 핵심이 되는 단계는 HIV의 RNA 게놈을 주형으로 DNA가 만들어지는 과정이다. 역전사효소 억제제는 바로 이 단계를 저해하는 화학물질이다. 역전사효소 억제는 크게 기질인 뉴클레오타이드/뉴클레오사이드와 화학적으로 유사한 유사체로 이루어진 뉴클레오타이드/뉴클레오사이드 역전사효소 저해제(Nucleoside/nucleotide reverse-transcriptase inhibitors, NRTI, NtRTI)와 기질과의 반응 자리와는 다른 곳에 결합하여 효소 활성을 저해하는 비 뉴클레오사이드 역전사효소 저해제(Non-nucleoside reverse-transcriptase inhibitors, NNRTI)로 구분된다.
- 인테그라아제 저해제(Intergrase Inhibitors): 역전사효소에 의해서 DNA로 변환된 HIV의 DNA를 숙주세포의 지놈에 끼워넣는 기능을 하는 단백질인 인테그라아제를 저해하는 화합물이다.
- 단백질 분해효소 저해제(Protease inhibitors): HIV의 지놈에는 구조단백질을 코딩하는 Gag 유전자와 단백질 분해효소, 역전사효소, 지놈 안에 DNA를 융합시키는 인테그라아제를 코딩하는 Pol 유전자가 있다. 이들은 일단 하나의 단백질로 번역되어 만들어진 후에 단백질 분해효소에 의해서 절단되어 기능하는 단백질이 된다. HIV의 단백질 분해효소를 억제하면 바이러스 단백질 형성이 억제되게 된다.
즉 HIV의 생육 단계 중 여러 단계를 동시에 저해함으로써 최대한 바이러스의 증식을 억제하는 것이 HAART의 기본적인 개념이라고 하겠다. 물론 이러한 치료법이 바로 등장한 것은 아니고 HAART를 구성하는 여러가지 약물들은 독립적으로 개발이 진행되었고, 그 이후에 이들을 병용하는 ‘칵테일 요법’이 등장하였다. 일단 가장 먼저 개발에 들어간 역전사효소 억제제의 개발과정부터 알아보도록 하자.
최초의 HIV 항바이러스 요법제제 아지토티미딘
가장 최초로 사용된 아지도티미딘(azidothymidine, AZT)은 화학 구조로 쉽게 유추할 수 있듯이 DNA의 구성 물질인 티미딘(Thymidine)의 유사체이다. 인산기가 붙지 않은 뉴클레오시드(Nucleoside)인 AZT는 생체 내에서 인산기가 결합되어 뉴클레이타이드(Nucleotide)가 되고, RNA를 주형으로 DNA를 만드는 역전사효소의 활성자리에 결합하여 역전사효소를 저해시킨다.
AZT는 아직 HIV/AIDS의 위험이 본격화되기 한참 전인 1964년 디트로이트 암 연구소(현재의 바바라 앤 카마로스 암 연구소 Barbara Ann Karmanos Cancer Institute)에 근무하던 연구자인 제롬 호위츠(Jerome P. Horwitz, 1919-2012)에 의해 처음 합성되었다[1]. 호위츠는 DNA 합성의 전구체인 티미딘의 유사체를 만들면 DNA 합성을 저해하여 암세포의 증식을 억제할 것이라는 가설하에 티미딘의 유사체인 AZT 및 여러 종류의 뉴클레오사이드 유사체(현재 레트로바이러스 저해제로 역사 사용되는 스타뷰딘 Stavudine 및 잘시타빈 Zalcitabine)를 합성하였다. 그러나 그의 기대와는 달리 AZT는 암 모델 동물에서 전혀 항암 효과를 내지 못했다. 그리하여 AZT는 기대한 효과를 내지 못한 수많은 다른 화합물들과 함께 잊혀져 있었다.
1974년, 암을 유발하는 레트로바이러스에 대한 연구가 활발하던 시절 독일의 막스 플랑크연구소의 연구자들이 AZT가 마우스 백혈병 바이러스(Murine Leukemia Virus)의 일종인 프렌드 바이러스의 증식을 억제한다는 것이 발견되었다[2]. 프렌드 바이러스 역시 레트로바이러스였고 이 결과는 레트로바이러스에 의해 유발될 수 있는 질병을 치료할 수 있다는 것을 암시하는 중요한 결과였다. 그러나 이전 연재에서 설명했듯이 인간에서 암을 일으키는 레트로바이러스는 거의 발견되지 않았으므로 이 발견 역시 크게 주목받지 않았다.
그러나 1980년대에 들어 HIV/AIDS의 문제가 심각해지며 HIV를 억제하는 항바이러스 요법에 대한 관심이 높아지면서 잊혀졌던 AZT 역시 재발견되었다. 지난 연재에서 알아본 것처럼 1983년 HIV가 처음 발견된 이후 곧 레트로바이러스 증식을 억제하는 화학물질을 찾는 연구가 바로 시작될 수 있었다. 특히 1970년대 미국 국립보건원 산하 국립암연구소(National Cancer Institute)에서 ’암과의 전쟁’에서 레트로바이러스 관련 연구가 많이 진행되었고 이를 억제하는 화합물을 찾기 위한 기반 기술들이 많이 개발되어 있던 상태였으므로 이러한 기반 기술을 이용하여 바이러스를 억제하는 물질의 탐색 또한 신속히 진행될 수 있었다.
1985년 NCI 연구자들은 HIV에 감염된 T세포 라인에 AZT를 처리하면 바이러스의 증식과 역전사효소 활성을 억제한다는 것을 발견하였다[3]. 이와 비슷한 시점에 버로우즈-웰컴(Burroughs-Wellcome, 지금의 GSK)사에서도 세포 수준에서 AZT의 항 HIV 활성을 확인하였다. 그렇다면 실패한 항암제였던 AZT가 어떻게 항레트로바이러스 약제로써 효과가 있을 수 있었을까? 1986년의 AZT를 이용한 생화학적 기전 연구에서 AZT는 세포 내의 DNA 중합효소에 비해 HIV의 역전사효소에 100배 이상 강한 친화력을 가진다는 것이 밝혀졌다[4].
세포 수준에서의 효과를 관찰한지 몇개월만에 신속하게 임상 1상이 시작되었다. 현대의 신약 개발에서는 실험동물 수준에서의 전임상 연구가 진행되어 효과와 독성을 면밀히 검증받고 임상에 들어가는 것이 일반적이지만, 당시의 HIV/AIDS에 대한 위협은 이런 절차를 대개 생략하고 환자 대상의 임상으로 바로 직행할 정도로 심각한 상태였다. 게다가 항 HIV 활성을 검증할 적절한 동물 모델 역시 없는 상황이었다. NCI와 듀크대학의 연구자들에 의해서 수행된 임상 1상의 결과는 1986년 발표되어 HIV 환자에게 심각한 부작용 없이 투여될 수 있었고 CD4 T세포의 숫자와 T세포 면역을 회복시킬 수 있다는 희망을 주게 하였다[5].
곧 이어 282명의 AIDS 환자를 대상으로 24주에 걸쳐서 250mg의 AZT 혹은 위약을 4시간에 한번씩 복용하는 이중맹검 임상시험이 진행되었다. 그 결과 위약군에서는 19명의 환자가 사망한 반면 AZT 복용군에서는 1명의 환자가 사망하였으며, T세포의 숫자나 T세포 면역성이 회복되는 것이 확인되었다[6]. 이러한 임상 시험 결과에 기반하여 FDA는 1987년 3월 AZT의 HIV 감염자 대상으로의 사용이 허가되었고, AZT는 지도부딘(Zidobudine)이라는 이름으로 HIV 감염자 치료에 사용되기 시작하였다. 시험관 수준에서 AZT의 활성을 확인한 시점부터 AZT의 FDA승인까지는 불과 25개월밖에 걸리지 않았으며 이러한 전례없는 속도는 당시의 AIDS에 대한 공포와 빠른 시간 내에 대책을 내놓아야 한다는 FDA의 방침 때문이었다.
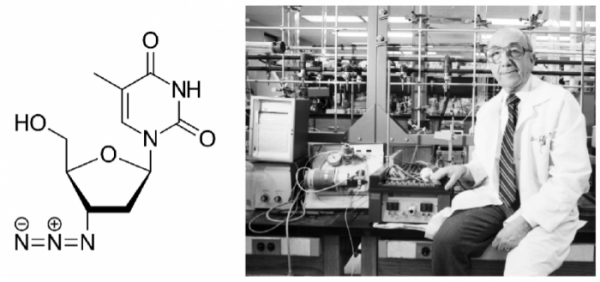
▲그림 1. (좌) 최초의 HIV 감염자 치료에 사용된 아지도티미딘 (azidothymidine, AZT)의 화학 구조 (우) AZT의 개발자 제롬 호위츠 (Jerome Horwitz). AZT는 DNA의 티민 뉴클레오타이드의 유사체로써 세포 내에 들어가 인산화되어 뉴클레오타이드가 된 이후 역전사효소를 저해한다. 원래 항암제로써 암세포의 DNA 합성을 저해하려는 목적으로 탄생한 AZT는 항암 효과가 나타나지 않아 잊혀진 물질이었으나 개발된지 20년 후에 역전사효소를 강하게 저해한다는 것이 발견되어 항바이러스제재로써 부활하게 되었다.
AZT 내성 바이러스의 출현과 비 뉴클레오사이드 역전사효소 저해제의 발굴
그러나 AZT가 1987년부터 HIV 감염자 치료에 사용되기 시작한지 얼마 지나지 않아 AZT에 내성을 가지는 바이러스가 쉽게 출현한다는 것이 발견되었다. 1989년 버로우즈 웰컴의 연구자들은 AZT에 내성을 가지는 HIV 바이러스는 HIV의 역전사효소 영역에 생긴 돌연변이에 의해서 발생한다는 것을 보고했다[7]. HIV의 역전사효소는 DNA 복제에서 돌연변이를 훨씬 많이 만들어 내고, 역전사효소 억제제를 장기 복용하는 환자에게서 살아남는 HIV 바이러스는 AZT가 역전사효소에 붙는 것을 방해하는 돌연변이를 가지도록 선택되는 것이다. 곧 이어 여러 종류의 AZT에 내성을 갖는 바이러스가 발견되었다.
이러한 것은 한 종류의 약물로써 HIV와 같이 빈번한 돌연변이를 보이는 바이러스를 상대하는 것은 역부족이라는 것을 의미한다. HIV의 증식을 효과적으로 억제하기 위해서는 다양한 기전에 의거한 복수의 화합물이 필요하다는 것에 눈을 뜬 연구자들은 다른 화합물의 발굴에 뛰어들었다.
1989년 HEPT(1-[(2-hydroxy-ethoxy) methyl]-6-phenylthiothymine)라는 화합물이 레트로바이러스 복제를 저해한다는 것이 발견되었다[8]. 곧 이어 이와는 전혀 다른 구조를 가진 TIBO(tetrahydroimidazo- [4,5,1-jk-[1,4]benzodiazepin-2(1H)-one)라는 물질 역시 발굴되었다[9]. 이들은 화학적으로 공통적인 구조를 가지지 않았지만 역전사효소를 저해하는데, AZT와 같이 뉴클레오타이드의 결합자리에 붙어서 뉴클레오타이드가 추가되는 것을 방해하는 것이 아니라, 뉴클레오타이드가 추가되는 위치와는 약 10 옹스트롬 떨어진 위치에 붙어서 역전사효소의 활성을 억제하는 비 뉴클레오사이드 역전사효소 저해제(Non-nucleoside reverse-transcriptase inhibitors, NNRTI)의 최초 선도물질이었다. 1996년 TIBO로부터 유래된 네비라핀(Nevirapine)이라는 물질이 비 뉴클레오사이드 역전사효소 저해제로는 최초로 FDA의 승인을 얻었으며 1997년에는 델라비딘(delavirdine)이, 1998년에는 에파비레즈(efavirez)가 뒤이어 승인을 얻었다.
단백질 분해효소 억제제와 ‘칵테일 요법’ 의 등장
앞에서 설명한 것처럼 HIV의 복제 사이클에서 HIV가 가지고 있는 단백질 분해효소에 의한 Gag, Pol 단백질의 절단은 바이러스 복제에 필수적이다. 그렇다면 HIV의 단백질 분해효소의 특성을 보다 정확히 파악하면 단백질 분해효소를 억제할 수 있는 화합물을 만드는 데 보탬이 될 수 있을 것이다.
1989년, HIV의 단백질 중 최초로 HIV의 단백질 분해효소의 3차원 구조가 규명되었다[10]. 이렇게 규명된 HIV의 단백질은 이합체(Dimer)로 두개의 서브유니트가 결합되어 존재하고 있었고 효소의 활성 자리는 아스파르트산(Aspartic Acids)이 존재하는 아스파르트산 단백질 분해 효소(Aspartic Protease)였다. 비교적 이전에 많이 되어 있던 아스파르트산 단백질 분해효소와 비슷한 성질을 가진 단백질이었고, 규명된 단백질 구조 정보를 이용하여 이를 저해할 수 있는 화합물이 디자인되었다. 이러한 단백질 분해효소 저해제는 단백질 분해효소의 원래 기질인 펩타이드 서열과 유사하되 분해가 되지 않도록 변형된 펩타이드 유사체(Peptidomimetics)계열의 물질이 많았다.
1990년 스미스클라인(Smithkline), 업존(Upjohn), 호프만 라 로슈(Hoffman-La Roche) 등 복수 제약회사의 연구진들은 각각 이들이 개발한 단백질 분해효소를 저해하는 펩타이드 유사체가 세포 내에서 HIV의 증식을 억제하는 것을 확인하였다[11].
이중 가장 빠르게 승인을 얻은 화합물은 호프만 라-로슈가 개발한 사퀴나비르(Saquinavir)라는 약물이었다. 사퀴나비르는 HIV 단백질 분해효소가 HIV 의 gag, Pol 단백질에 있는 페닐알라닌-프롤린의 사이, 혹은 타이로신-프롤린 사이를 절단한다는 것에 착안하여 이들 기질의 중간 전이 상태(Transition state)와 비슷하게 디자인된 화합물이다. 아스파르트산 단백질 분해효소는 생체에도 많이 존재하지만, 페닐알라닌과 프롤린 사이를 절단하는 단백질 분해효소는 별로 없다는 것을 고려하면 생체 내에 있는 다른 단백질 분해효소는 그다지 저해하지 않고 HIV 단백질 분해효소를 특이적으로 저해할 수 있을 것으로 생각되었다.
1996년 사퀴나비르와 기존의 역전사효소 저해제인 지도부딘과 잘시타빈(Zalcitabine)과 병용하여 효과를 보는 임상 시험 결과가 보고되었다[12]/ 사퀴나비르(매일 1800mg)와 지도부딘(매일 600mg), 잘시타빈(매일 2.25mg) 3가지의 약물을 복용하는 것과 지도부딘, 사퀴나비르, 혹은 지도부딘, 잘시타빈의 2가지 약물을 복용하는 그룹을 24주 동안 관찰하였다. 그 결과 3가지의 약물을 병용한 그룹이 CD4+ T세포의 숫자, HIV 의 증식 등 모든 지표에서 두 가지 약물을 사용한 것보다 우월하다는 것이 입증되었다. 1995년 12월 FDA는 사퀴나비르를 지도부딘 및 잘시타빈과 병용하여 사용하도록 승인하였으며 사퀴나비르는 인비레이즈(Invirase)라는 상표명으로 판매되기 시작하였다. 사퀴나비르는 최초로 승인된 HIV 단백질 분해효소 저해제였다. 곧 이어 애브비(AbbVie Inc)의 리토나비르(Ritonavir, 상품명 Norvir), 머크(Merck)의 인디나비르(Indinavir, 상품명 Crixivan) 등이 연이어 승인을 받았다. 특히 리토나비르의 경우 HIV 단백질 분해효소 저해제 능력도 있지만 간에서 약물을 분해하는 효소인 사이토크롬 P450 3A4를 저해하는 특성을 가지고 있었다. 이러한 특성은 혈중에서 다른 약물의 지속 기간을 늘림으로써 약물의 효과를 극대화하는 예상치 않은 특성을 가지기도 하였다.
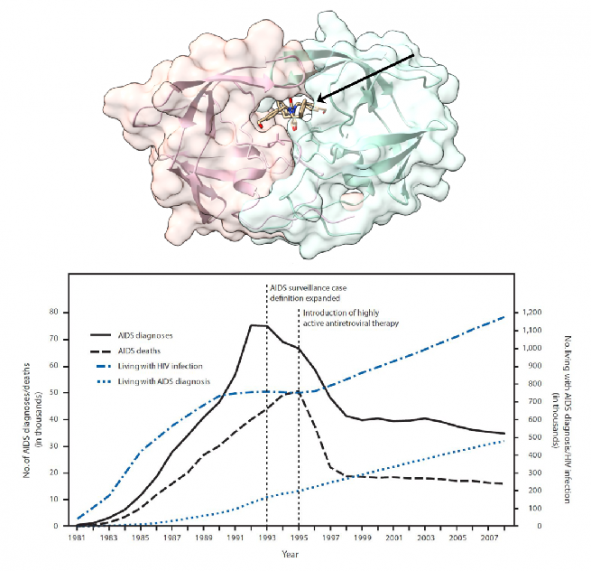
▲그림 2. HIV 단백질 분해효소에 결합되어 있는 단백질 분해효소 저해제. HIV 단백질 분해효소는 HIV 유래 단백질로는 최초로 단백질 구조가 규명되었으며 규명된 단백질 구조에 기반하여 디자인된 약물은 HIV 단백질 프로세싱을 저해하여 HIV 증식을 억제한다. HIV 단백질 분해효소 저해제는 다른 약물과 함께 HAART의 핵심 구성요소로써 HIV 감염자의 치료에 1995년경부터 사용되기 시작하였으며, HAART 등장후에 AIDS에 의한 사망자는 급격히 줄어들게 되었다.
단백질 분해효소 저해제의 등장과 역전사효소 저해제와의 병용 사용은 HIV 감염자 치료에서 획기적인 전기가 되었다. 단백질 분해효소 저해제 이전에 사용되던 역전사효소 저해제의 경우 여러 약물의 복합 치료에도 불구하고 바이러스를 오랜 기간동안 억제하는 것은 어려웠고, 결국 환자는 면역력 악화로 사망하는 경우가 많았다[13]. 그러나 단백질 분해효소 저해제와 두 종의 역전사효소 저해제를 병용한 임상 사례들이 1996년에서 1997년 앞다투어 보고되고[14], 이렇게 복수의 약제를 사용하는 치료법이 기존의 치료법에 비해서 현저히 좋은 효과를 보여주게 되면서 단백질 분해효소 저해제와 복수의 역전사효소 저해제를 동시에 투여하는 것이 표준적인 HIV 감염자의 치료법으로 대두되었다. 이 치료법은 고활성 항레트로바이러스 치료법(highly active antiretroviral treatment. HAART)이라는 명칭으로 불리게 되었으며 대중들에게는 ‘칵테일 요법’(Cocktail Therapy)로 알려지게 되었다.
그렇다면 HAART의 등장은 HIV 감염자의 생존 확률을 어느 정도 개선시켰을까? HAART 가 도입된지 얼마 안되었던 1999년의 추적 연구에 의하면 1994년에는 HIV 감염자 100명당 23.7건의 면역력 약화에 의한 질병이 발생하였고 20.2명이 사명하였던 반면 HAART 도입 이후인 1998년에는 질병 발생이 100인 중 14건으로 줄었고 사망자는 100명당 8.4 건으로 줄었다[15]. HAART가 표준 치료방법으로 등장한 지 얼마 안된 시점에서도 이미 가시적인 효과가 나타나기 시작한 것이다.
HAART에 사용되는 약물들이 지속적으로 개선되고 보다 효율적인 투약법들이 연구되기 시작하면서 HIV 감염자의 생존 확률은 급격하게 개선되었다. 2017년의 스위스의 HIV 감염자를 대상으로 한 추적 연구에서 HAART 등장 이후에 HIV 에 대한 치료법이 HIV 감염자의 생존 확률을 얼마나 높였는지를 명확하게 볼 수 있다[16]. AZT에 의한 HIV 감염자 치료가 처음 시작되었던 1988-1991년에 20세의 HIV 감염자의 평균 기대 수명은 11.8년이었다. 그러나 그동안 개선된 약제를 4종 이상 사용하는 치료법을 받는 2006-2013년의 20세 HIV 감염자의 기대 수명은 54.9 년에 달했다. 이러한 수치는 교육과 생활 수준에 비해서 더 올라가서 상대적으로 치료를 받을 확률이 높은 고학력자 HIV 감염자의 기대 수명은 60년으로 HIV 비감염자의 61.5년과 큰 차이가 없었다. 한마디로 이제 HIV 에 감염되어도 지속적인 치료와 관리를 받으면 정상인과 그다지 차이 없는 수명을 누릴 수 있는 시대가 된 것이다. 1980년대 중반까지만 하더라도 인류의 종말을 가져올 수 있는 ‘제 2의 흑사병’ 으로 인식되던 HIV/AIDS는 오늘날은 당뇨나 고혈압처럼 관리 가능한 만성 질환 쪽에 가깝게 변한 것은 전적으로 HAART 등의 항 바이러스 요법의 발전 덕이라고 봐야 하겠다.
물론 HIV와 AIDS의 위협이 이제 완전히 없어졌다고 이해하는 것은 곤란하다. 아직도 아프리카 등 공중보건과 제대로 된 치료를 받기 어려운 지역에서는 AIDS는 심각한 공중보건적 문제로 남아 있으며, 아직도 HIV/AIDS를 예방할 수 있는 백신의 개발은 요원한 실정이다. 다음 연재에서는 왜 HIV에 대한 백신 개발의 어려움과 이를 극복하기 위해서 어떤 연구가 진행되어 왔는지에 대한 이야기를 다루며 HIV/AIDS에 대한 이야기를 마무리하도록 한다.
참고문헌
Horwitz, J. P., Chua, J., & Noel, M. (1964). Nucleosides. V. The Monomesylates of 1-(2'-Deoxy-β-D-lyxofuranosyl) thymine The Journal of Organic Chemistry, 29(7), 2076-2078.
Ostertag, W., Roesler, G., Krieg, C. J., Kind, J., Cole, T., Crozier, T., ... & Dube, S. (1974). Induction of endogenous virus and of thymidine kinase by bromodeoxyuridine in cell cultures transformed by Friend virus. Proceedings of the National Academy of Sciences, 71(12), 4980-4985.
Mitsuya, H., Weinhold, K. J., Furman, P. A., St Clair, M. H., Lehrman, S. N., Gallo, R. C., ... & Broder, S. (1985). 3'-Azido-3'-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proceedings of the National Academy of Sciences, 82(20), 7096-7100.
Furman, P. A., Fyfe, J. A., St Clair, M. H., Weinhold, K., Rideout, J. L., Freeman, G. A., ... & Mitsuya, H. (1986). Phosphorylation of 3'-azido-3'-deoxythymidine and selective interaction of the 5'-triphosphate with human immunodeficiency virus reverse transcriptase. Proceedings of the National Academy of Sciences, 83(21), 8333-8337.
Yarchoan, R., Weinhold, K., Lyerly, H. K., Gelmann, E., Blum, R., Shearer, G., ... & Markham, P. (1986). Administration of 3'-azido-3'-deoxythymidine, an inhibitor of HTLV-III/LAV replication, to patients with AIDS or AIDS-related complex. The Lancet, 327(8481), 575-580.
Fischl, M. A., Richman, D. D., Grieco, M. H., Gottlieb, M. S., Volberding, P. A., … Laskin, O. L. (1987). The Efficacy of Azidothymidine (AZT) in the Treatment of Patients with AIDS and AIDS-Related Complex. New England Journal of Medicine, 317(4), 185–191. doi:10.1056/nejm198707233170401
Larder, B., & Kemp, S. (1989). Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science, 246(4934), 1155–1158. doi:10.1126/science.2479983
Baba, M., Tanaka, H., De Clercq, E., Pauwels, R., Balzarini, J., Schols, D., ... & Miyasaka, T. (1989). Highly specific inhibition of human immunodeficiency virus type 1 by a novel 6-substituted acyclouridine derivative. Biochemical and biophysical research communications, 165(3), 1375-1381.
Debyser, Z., Pauwels, R., Andries, K., Desmyter, J., Kukla, M., Janssen, P. A., & De Clercq, E. (1991). An antiviral target on reverse transcriptase of human immunodeficiency virus type 1 revealed by tetrahydroimidazo-[4,5,1-jk] [1,4]benzodiazepin-2 (1H)-one and -thione derivatives. Proceedings of the National Academy of Sciences, 88(4), 1451–1455. doi:10.1073/pnas.88.4.1451
Navia, M. A., Fitzgerald, P. M., McKeever, B. M., Leu, C. T., Heimbach, J. C., Herber, W. K., ... & Springer, J. P. (1989). Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature, 337(6208), 615.
McQuade, T., Tomasselli, A., Liu, L., Karacostas, V., Moss, B., Sawyer, T., … Tarpley, W. (1990). A synthetic HIV-1 protease inhibitor with antiviral activity arrests HIV-like particle maturation. Science, 247(4941), 454–456. doi:10.1126/science.2405486 ; Meek, T. D., Lambert, D. M., Dreyer, G. B., Carr, T. J., Tomaszek, T. A., Moore, M. L., … Petteway, S. R. (1990). Inhibition of HIV-1 protease in infected T-lymphocytes by synthetic peptide analogues. Nature, 343(6253), 90–92. doi:10.1038/343090a0;Roberts, N. A., Martin, J. A., Kinchington, D., Broadhurst, A. V., Craig, J. C., Duncan, I. B., ... & Krohn, A. (1990). Rational design of peptide-based HIV proteinase inhibitors. Science, 248(4953), 358-361
. Collier, A. C., Coombs, R. W., Schoenfeld, D. A., Bassett, R. L., Timpone, J., Baruch, A., ... & Friedman, H. M. (1996). Treatment of human immunodeficiency virus infection with saquinavir, zidovudine, and zalcitabine. New England Journal of Medicine, 334(16), 1011-1018.
Moore, R. D. (1996). Natural History of Opportunistic Disease in an HIV-Infected Urban Clinical Cohort. Annals of Internal Medicine, 124(7), 633. doi:10.7326/0003-4819-124-7-199604010-00003
Hammer, S. M., Squires, K. E., Hughes, M. D., Grimes, J. M., Demeter, L. M., Currier, J. S., ... & Chodakewitz, J. A. (1997). A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. New England Journal of Medicine, 337(11), 725-733; Gulick, R. M., Mellors, J. W., Havlir, D., Eron, J. J., Gonzalez, C., McMahon, D., ... & Emini, E. A. (1997). Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. New England Journal of Medicine, 337(11), 734-739.
Moore, R. D., & Chaisson, R. E. (1999). Natural history of HIV infection in the_era of combination antiretroviral therapy. Aids, 13(14), 1933-1942.
Gueler, A., Moser, A., Calmy, A., Günthard, H. F., Bernasconi, E., Furrer, H., ... & Zwahlen, M. (2017). Life expectancy in HIV-positive persons in Switzerland: matched comparison with general population. AIDS (London, England), 31(3), 427.







