기사본문
[BioS 레터]Bioinformatics의 응용..WGS 암변이 분석
입력 2020-11-24 10:32 수정 2020-11-24 10:32
이의정 객원연구원
2010년, 캐나다에서 한 연구팀이 전체 게놈(Whole Genome)분석에 따른 임상 응용이 가능한지에 대한 시도가 있었다. 이 연구팀은 혀의 선암(Adenocarcinoma) 조직의 유전체를 서열화하고 증폭하여, RET 종양 유전자가 잠재적인 원인이라는 것을 확인하였다. 이 분석을 통해 RET 단백질을 목표로하는 Kinase억제제를 사용함으로써, 해당 암환자를 위한 개인화된 치료 접근법을 이끌어 낼 수 있었다.
차세대염기서열분석법(Next-Generation Sequencing, NGS)에 대해 많이 들어보았을 것이다. 이는 유전체를 무수히 많은 조각으로 자른 뒤, 각각의 염기들을 읽어내어 그 서열을 데이터로 만든다. 이러한 유전체 데이터를 여러 생물 정보학적 분석방법들을 통해 빠르게 해독하고 분석하는 과정을 거친다. 이 중 전장 유전체 분석(Whole Genome Sequencing, WGS)은 하나의 종에 대한 유전정보 전체를 분석하는 방법으로, 최근 빠른 기술 발전과 가격이 낮아지면서 연구가 활발히 진행되고 있다. 이 분석은 비암호화 영역의 유전체(Noncoding Region; 단백질 코딩 지역 이외의 유전체 부위)도 함께 분석이 가능하다는 이점이 있어, 이제까지 비코딩 영역에서의 제한되었던 정보에 대한 해결책을 가져다주었다.
WGS 분석은 암과 관련된 변이(Mutation)들의 변화를 연구하고, 아직 알려지지 않은 변이에 대한 정보와 분자 치료 및 질병 예측을 위한 연구를 위해 사용될 수 있다. 암은 본질적으로 체세포 돌연변이의 축적과 함께 진화하고 진행되는 유전적인 질병 중 하나로, Copy Number Alterations (CNA)와 Structural Variants(SV), 후성유전학적 변이들을 나타내기도 한다. 예를 들어, 암 발병 원인들 중에서 암 억제 유전자인 RB1, TP53, APC에 대한 변이는 Loss‐of‐heterozygosity(LOH) 분석을 통해 확인이 가능하였고, 종양 활성 유전자인 HER2/ERBB2와 MYC은 Copy Number 분석을 통해 발견할 수 있었다.
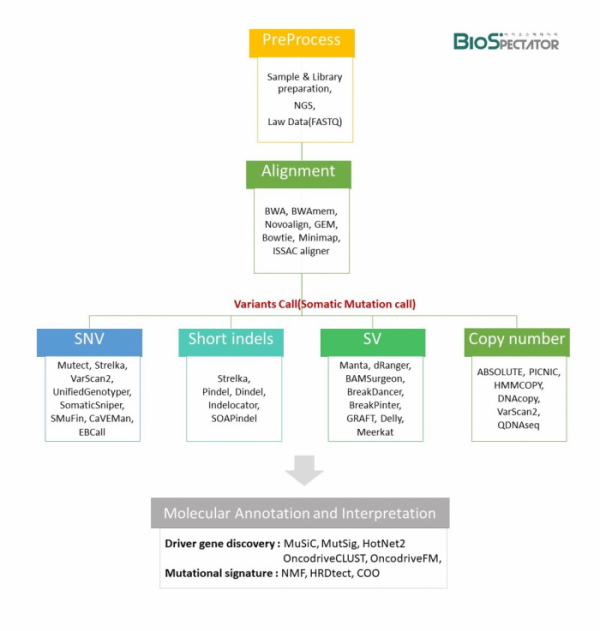
암 연구를 위한 WGS분석은 여러 분석 프로그램과 단계를 거친다. 우선, 비교가능한 정상세포와 암세포로부터 유전체 샘플을 추출하여 시퀀싱을 위한 Library를 제작한다. NGS로부터 생산된 염기서열 데이터는 FASTQ파일로 저장되고, 이는 BWAmem과 같은 Aligner 프로그램에 의해 인간 게놈 참조 염기서열(hg19 또는 hg38)을 따라 정렬되면서 BAM 파일을 생성한다. 이 후, 다양한 프로그램들을 사용하여 Single Nucleotide Variants(SNV), Short Indels, CNA, SV과 같은 체세포 변이들을 찾아내면서 암세포에만 특이적으로 존재하는 변이들을 확인하고 정상 세포와 암세포에서의 Variant Allele Fractions(VAF) 비교에 대한 알고리즘을 거치게 된다. 체세포 변이 발견은 발굴한 유전자 변이에 대한 특징을 주석처리하는 Annotation과정을 거친다. dbSNP, 1000 Genome Project, Exome Sequencing Project(ESP), ExAC, ClinVar 등의 데이터베이스를 통해 기존에 알려진 변이들과의 상관관계 및 단백질 서열 결과, 질병 등에 대한 정보를 추가한다. Oncotator와 CRAVAT는 암특이적인 주석을 제공하는 도구로 널리 사용되고 있으며, 암과 관련된 정보들을 제공한다. 이 알고리즘들은 정확한 변이를 찾기 위해 현재도 계속 다양한 프로그램들이 개발되고 있다.
Whole Exome Sequencing(WES) 분석은 단백질 코딩 영역에서의 많은 변이들이 발견하는데 많은 발전을 이뤄냈지만, 그에 비해 비코딩 영역에서는 정보들은 아직 덜 알려져 있었다. WGS 분석 가격이 낮아지고 분석 환경이 개선되면서 프로모터(Promoter)나 인핸서(Enhancer)와 같은 비코딩 영역에서도 암에 영향을 줄 수 있는 변이들이 발견되고 있다.
Rheinbay 연구팀은 360개의 유방암샘플과 정상샘플을 비교하여 예상보다 돌연변이가 많거나, 비정상적인 변이가 군집되어 있는 프로모터를 찾는 방법으로 6개의 프로모터(TBC1D12, LEPROTL1, ZNF143, RMRP, ALDOA, FOXA1)에서 변이가 일어나는 것을 확인하였다. 이 프로모터들의 변이에 대한 정확한 역할을 알기 위해 Luciferase reporter assays, Pull-down assay, EMSAs 등의 다양한 분자생물학적 실험들이 진행되었고, 이 결과로 Estrogen Receptor-positive 유방암 유도물질인 FOXA1(Forkhead Box A1) 프로모터 위치에서 변이(G>A)를 찾아낼 수 있었다. 이 변이는 FOXA1의 과발현을 이끌어 E2F와의 상호작용 증가시키고 결국 암세포를 증가시켰다. 이 연구는 FOXA1 변화를 통해 종양 진행성을 파악하고 메커니즘을 인식하여 호르몬 치료를 받는 유방암 환자들에게 도움을 줄 수 있는 가능성을 알려주었다.
바이러스나 박테리아 감염은 만성 염증을 일으키고 이는 암 발생에도 강력한 원인으로 작용한다. 우리가 흔히 알고 있듯이, Hepatitis B virus(HBV)와 Hepatitis C virus(HCV) 감염은 간암을 일으키고 Human papillomavirus(HPV) 감염은 자궁경부암을, Helicobactor pylori와 Epstein-Barr virus (EBV) 감염은 위암을 유발시킨다. Sung WK 연구팀은 간암(Hepatocellular Carcinomas, HCCs) 세포와 정상세포를 시퀀싱하고 비교 분석하여, 염색체 불안정성이 높은 HBV Breakpoint 위치에서 Copy Number Variation(CNV)가 눈에 띄게 증가된 것을 증명하였다. 또한, 암세포에서 과발현된 TERT, MLL4, CCNE1 유전자가 세포안에서의 HBV 변화 과정과 관련이 있다는 것도 확인하였다. 또 다른 예로 Ojesina 연구팀은 자궁경부암세포와 정상세포를 비교하는 WGS 분석과 WES, RNA sequencing 등의 여러 분석과 실험을 통해 총 1만7,795개의 체세포 변이와 Chromosome 8q24를 포함한 여러 HPV의 통합 위치들을 찾아내었고, HPV 융합과 주변 유전자의 발현 증가 사이의 연관성 및 Mitogen-Activated Protein Kinase 1(MAPK1)의 E322K(Missense mutation, G>A) 변이도 확인하였다.
이 외에도 재배열(Rearrangements)과 SV 변이는 특정 암의 진단도구로 쓰이기도 하며, CNA도 가장 흔한 암 특징으로 종양 억제 유전자의 비활성화 또는 종양 유전자를 활성화시키기도 한다. 암에 대한 WGS분석은 WES와 달리, WGS는 많은 암에서 수천 개의 체세포 변이들을 찾아내고 비코딩 영역의 연구도 가능하다는 이점을 갖는다.
최근 WGS 분석은 NGS의 가격 하락과 아마존 및 구글 클라우드 등의 빅데이터 사용가능한 환경 개선으로 대규모 전장 유전체를 효율적으로 분석할 수 있게 되어 더욱 활발해지고 있다. 하지만, 분석 플랫폼들에 대한 데이터 퀄리티 및 정확성 등의 한계점들은 지속적인 발전을 이끌어내는 이유로 남아있다. 이를 극복하기 위해 많은 기술 개발과, RNA-Seq, 후성유전학, 면역유전학 및 임상병리학적 정보와 같은 통합된 대규모 WGS 데이터의 분석과 연구들이 진행 중이다. 앞으로의 더욱 발전된 기술들을 통해 개개인의 암 환자들의 특성에 따라 치료와 완치가 가능한 날을 기대해 본다.
Reference
1. Jones, S. J. et al, Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors, Genome Biol, 2010, 11, Article number: R82
2. Eric Y. Zhao et al, Whole-Genome Sequencing in Cancer, Cold Spring Harbor Perspectives in Medicine, 2020; 10.1101/cshperspect.a034579
3. Elaine R. Mardis and Richard K. Wilson, Cancer genome sequencing: a review, Hum Mol Genet. 2009; 18(R2): R163–R168.
4. Hidewaki Nakagawa and Masashi Fujita, Whole genome sequencing analysis for cancer genomics and precision medicine, Cancer Sci. 2018; 109(3): 513–522
5. OU Sezerman et al, Bioinformatics workflows for genomic variant discovery, interpretation and prioritization, Bioinformatics Tools for Detection and Clinical Interpretation of Genomic Variations, 2019; https://doi.org/10.5772/intechopen.85524
6. Rheinbay E et al, Recurrent and functional regulatory mutations in breast cancer, Nature, 2017; 547:55-60.
7. Ojesina AI et al, Landscape of genomic alterations in cervical carcinomas, Nature, 2014; 506:371-375.




![[창간기획]이뮤노반트로 본 '신약개발' 3가지 메시지](https://img.etoday.co.kr/crop/268/200/2347309.jpg)

![[인사]롯데바이오, '암젠 출신' 한재준 CTO 영입](https://img.etoday.co.kr/crop/268/200/2351192.jpg)
