기사본문
CHMP, 사노피 '듀피젠트' 만성비부비동염 '승인 권고'
입력 2019-09-24 15:06 수정 2019-09-24 15:06
바이오스펙테이터 이승환 기자

사노피(Sanofi)와 리제네론(Regeneron)이 공동개발한 IL-4, IL-13 항체 ‘듀피젠트(Dupixent, 성분명: dupilumab)’가 유럽에서 비용종을 동반한 중증 만성비부비동염(chronic rhinosinusitis with nasal polyposis, CRSwNP) 치료제로 판매되는 것에 청신호가 켜졌다.
유럽의약품기구(EMA)의 산하 기관인 약물사용자문위원회(CHMP)는 지난 19일(현지시간) ‘듀피젠트+모메타손 푸로에이트(mometasone furoate)’ 병용요법을 비용종을 동반한 중증 만성비부비동염 환자에게 투여하는 것에 대해 유럽연합 집행위원회(EC)의 판매승인을 권고하는 긍정의견을 채택했다.
CHMP은 의약품의 안전성, 유효성 등을 평가해 판매승인 권고 또는 거절 의견을 EC에 전달한다. CHMP의 긍정의견을 받은 의약품은 EC의 심사를 거쳐 3개월 안에 최종 승인을 얻게 된다.
비용종을 동반한 중증 만성비부비동염 환자에게 듀피젠트 투여 시, 코르티코스테로이드(corticosteroid)제인 모메타손 푸로에이트를 비강 흡입형태로 함께 투여한다. IL-4, IL-13에 결합해 제2형 도움T세포(type 2 helper T cell, Th2)를 감소시키는 듀피젠트는 코르티코스테로이드와 병용해 아토피 피부염, 천식 환자의 과도한 면역반응을 억제한다.
CHMP는 24주간 276명의 환자가 참여한 임상3상(SINUS-24, NCT02912468), 52주간 448명의 환자가 참여한 임상3상(SINUS-52, NCT02898454) 2건의 결과를 근거로, EC에 승인 권고 의견을 전달했다. 두 임상시험에서 ‘듀피젠트+모메타손 푸로에이트’ 병용요법 투여그룹은 ‘듀피젠트 위약+모메타손 푸로에이트’ 위약그룹과 비교했다. 두 임상시험에서 1차 종결점으로 설정된 비충혈(nasal congestion), 비폐색(nasal obstruction), 비용종(nasal polyps), 2차 종결점으로 설정된 부비동 혼탁(sinus opacification), 후각 상실(loss of smell) 증상은 투여그룹이 위약그룹보다 개선된 것으로 나타났다. 투여그룹의 코르티코스테로이드를 지속해서 투여해야하는 환자 비율은 위약그룹 대비 74% 감소했으며, 부비동 수술이 필요한 환자 비율은 위약그룹 대비 83% 감소했다.
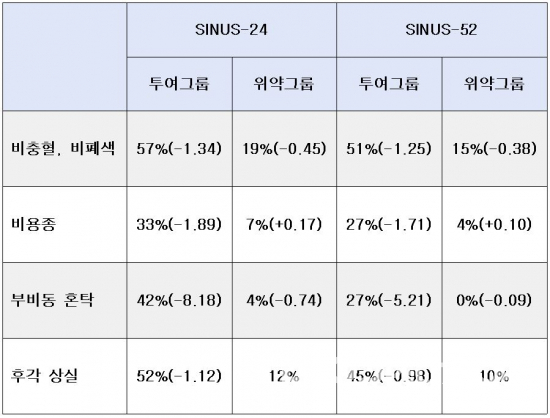
▲SINUS-24, SINUS-52 임상시험에서 증상 개선도 및 기준점 대비 최소제곱법 비교 결과, 후각 상실의 최소제곱법은 위약그룹 대비 비교(바이오스펙테이터 정리)
사노피는 EC가 10월 중에는 듀피젠트 승인 여부를 결정할 것으로 예상하고 있다. 듀피젠트가 EC로부터 승인받게 되면 아토피 피부염(atopic dermatitis), 천식(asthma)에 이어, 비용종을 동반한 중증 만성비부비동염을 세 번째 적응증으로 유럽에서 판매할 수 있게 된다. 듀피젠트는 지난 6월 미국 식품의약국(FDA)으로부터 비용종을 동반한 중증 만성비부비동염 치료제로 미국 내 판매를 승인받았다.
한편, 셀트리온의 램시마SC(성분명: infliximab)도 같은 날 CHMP로부터 류마티스 관절염(rheumatoid arthritis) 환자에게 투여할 수 있도록 승인 권고 의견을 받았다. 셀트리온은 램시마SC에 대한 유럽 판매승인 신청서를 작년 11월 EMA에 제출했다. CHMP의 승인 권고에 따라 램시마SC의 유럽 판매승인 여부는 늦어도 올해 말에는 알 수 있게 됐다.
다만 CHMP의 승인 권고가 반드시 EC의 승인으로 이어지는 것은 아니다. 대웅제약은 지난 4월 CHMP로부터 보툴리눔톡신제 ‘나보타(성분명: Clostridium botulinum toxin type A)’를 미간 주름 개선을 위해 투여하는 것에 대해 긍정의견을 받았다. 하지만 지난 6월 EC가 보완자료를 요구하면서 판매승인 결정이 7월 이후로 연기됐다. 이에 대웅제약은 EC가 요구한 자료는 판매승인에 영향을 줄 자료가 아니라고 밝혔다. 나보타는 지난 2월 FDA로부터 미국 내 판매를 승인받았는데, 나보타가 FDA의 심사를 거칠 때도 한차례의 자료보완 요구가 있었다. 대웅제약은 나보타의 유럽 판매승인에도 문제가 없을 것으로 전망한다.







